How much of human evolution was due to polygenic adaptation?

This has been an eventful week for those of us who study the dynamics of recent selection in humans. The most significant event was the publication of a paper describing genetic analysis of a long selection experiment in Drosophila. Although the experiment differs from most natural instances of selection in some important ways, the results give some very helpful corroboration that the recent human pattern of adaptive evolution was rapid and of an expected pattern for massive selection on many traits.
Meanwhile, Jonathan Pritchard and Anna Di Rienzo have a short review in the current Nature Reviews Genetics, discussing the idea that a large fraction of adaptive evolution may be difficult to find with current genetic evidence.
Their idea is that polygenic adaptations are unlikely to occur by successive “sweeps” of new adaptive mutations.
It seems likely to us that, as in traditional quantitative genetic models, many possibly even most adaptive events in natural populations occur by polygenic adaptation. Polygenic adaptation could allow rapid adaptive shifts, yet would often go undetected using conventional methods for detecting selection. Moreover, polygenic adaptation is qualitatively different from the models of adaptive substitutions that dominate the population genetics literature.
This is not a new idea, but Pritchard and Di Rienzo review it in a productive way, and the topic is worth some deeper thought…
An adaptive genetic substitution is often modeled as an episode of logistic growth. A new mutation, initially in a single copy, increases exponentially in numbers until it is very common in the population. After this point, it continues to increase in frequency up to fixation, but progressively slowly. The entire process takes hundreds or a few thousands of generations, which sounds like a long time but is actually very rapid compared to the deep genealogical histories of most genetic loci. The initial rapid increase in numbers carries a region of linked sequence along with the selected variant. This “hitchhiking” region is highly visible because of the co-association of nearby allelic variants. Thus, if such a “sweep” is ongoing, we should have little trouble finding it. In humans we’ve found a lot of them, which is a big piece of evidence for the rapidity of human evolution during the past 40,000 years.
But all that describes the dynamics of a single, strongly selected, mutation. What if a trait comes under selection, but the variation in the trait is explained not by a single gene, but by dozens or hundreds of genes? Pritchard and Di Rienzo outline such a scenario:
The key point is that we should expect such an adaptation to occur by small allele frequency shifts spread across many loci. As a hypothetical example, consider the adaptation of human height a trait for which there are probably hundreds of SNPs that each affect height by a few millimeters. Strong selection for increased height could be very effective, as height is extremely heritable. But at the level of individual SNPs, the effect of selection would be rather weak, exerting just a small upward pressure in favour of each of hundreds of 'tall' alleles. Suppose that at 500 SNPs, the tall alleles each increase the expected height of a person by 2 mm. Then, an average shift of just 10% in the population allele frequency of each tall allele would increase average height in the population by 20 cm (assuming that SNPs contribute additively). Although these numbers are hypothetical, they illustrate that, for a highly polygenic trait, a dramatic adaptive response could result from modest allele frequency changes at many loci. This model is different from classical sweep models. Most importantly, adaptation could occur without dramatic allele frequency changes and without adaptive fixation events.
But the description isn’t precisely what would happen in the case of selection on stature. Consider:
- It is true that alleles that already exist in the population provide the most immediate opportunity for change under directional selection. Any short-term phenotypic evolution we see is likely to be caused by changes in the frequency of standing variants.
- Some of the alleles that affect stature are constrained by their effects on other phenotypes. They might not change, even under directional selection on stature.
- Stature may be affected by hundreds of loci, but these do not account for equal proportions of the additive variance. Loci are subject to selection roughly in proportion to the additive variance in fitness they explain. Directional selection on stature will change the allele frequencies for a few loci quite a bit more quickly than most.
The distribution of effect sizes is fairly well known for stature in humans. For example, Park and colleagues this spring plotted the distribution of effect sizes for variants discovered by GWAS in 63,000 Europeans:
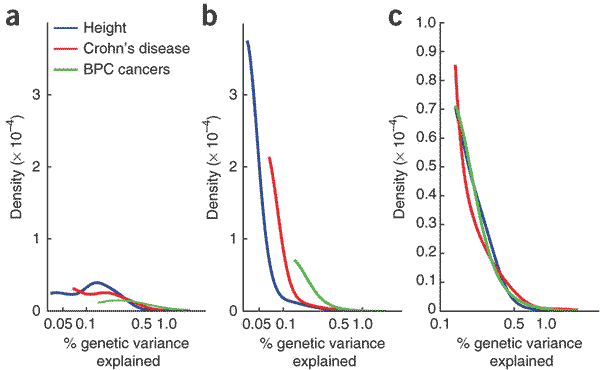
In the figure, (a) is based on observed loci – for stature, this includes 30 loci that reached significance in the GWAS without follow-up genotyping. There is a pretty severe ascertainment bias against small effect sizes, so curve (b) attempts to model the actual distribution correcting for ascertainment. Curve (c) is normalized to give the three conditions the same observed range.
You can see that if we suddenly started selecting for height, most of the genetic response would come from a very small proportion of the loci that explain the current additive variance. These would be the subset of loci in the large-effect-size tail of the distribution, excluding those that are constrained by their role in other phenotypes under selection.
- As an allele becomes common enough (going up toward fixation), the locus will account for less and less of the additive variance in fitness. To maintain the same response to selection, other alleles must pick up the slack. Over time, groups of different alleles will come into focus of selection, sort of like the “cover flow” feature of an iPod. Some alleles increase in frequency across a transient in the mid-frequency range, only to be gradually replaced by others. Most of the phenotypic change occurs as alleles cross rapidly from 40 to 60 percent or so.
- A few loci will be special. These account for an appreciable fraction of additive variance even though the favored allele is very rare. As they become common, these favored alleles change in frequency more and more rapidly, and account for more and more of the additive variance. They suck up the oxygen of selection. These alleles will look like a classic sweep.
- Over many generations, new mutations may occur that also have strong effects on the trait. They will follow the “special” pattern described in 5.
The question is how many loci of this type can we expect to exist? We all know that there are two patterns that could account for the heritability of traits like stature, where no common variants have very strong effects. Either the additive variance is spread across many rare variants with large effects, or instead across many common variants with small effects. Pritchard and Di Rienzo’s scenario accentuates the second of these – a small frequency change in many common variants with small effects.
But if even a small fraction of the additive variance is explained by a few rare variants with strong effects, these may cause most of the phenotypic change, and may look a lot like a standard selective sweep.
Pritchard and Di Rienzo note that the two options – a rapid sweep of one or a few locus, versus slight frequency changes in many loci – are not mutually exclusive. Most cases of directional selection on phenotypes may involve both patterns. If so, that will be very helpful, because we can use the easy-to-find sweeps to target analysis of harder-to-find frequency changes.
They sketch a strategy for examining the evolution of such traits.
One type of approach will be to identify phenotypes that may have undergone adaptive changes in particular environments, such as adaptations to cold climate, high altitude or novel ecological conditions. To dissect the genetic basis of such adaptations, one might collect phenotyped samples from closely related populations that have and have not experienced the selective pressure of interest and use GWA mapping to identify relevant quantitative trait loci (QTLs). Additionally, one would want to measure the extent of phenotypic adaptation estimated as the difference in average phenotype between the adapted and non-adapted populations when they are living under matched conditions (exact matching of conditions may be difficult in human studies). Then one could ask: what fraction of the phenotypic difference can be explained by alleles with large versus small frequency differences? Are the phenotypic effect sizes of QTLs with large allele frequency differences greater than those with subtle frequency shifts10? What fraction of the phenotypic difference cannot be explained by detected sweep signals or QTLs at all (and hence might result from the cumulative effect of many weak QTLs)? In another type of scenario, one might hypothesize that a particular aspect of the environment is an important selective factor (for example, climate or diet) but it is unclear what all the relevant phenotypes are. In this case, we might study adaptation by looking at sets of populations that have independently adapted to the same selective pressures. One type of signal would be alleles that show parallel frequency shifts in response to similar environmental pressures in distantly related populations (although this type of approach is unlikely to be powerful for alleles with very small effects).
These are exactly the kind of tests that we are working on here at Wisconsin. We have some pretty promising ideas, I think. If you’re on a dissertation grant panel, would you please give some money to my students who want to apply these approaches?
I mean, really, this is the best application of anthropology to develop new genetic approaches, rich in theory and in empirical evidence. Humans are the ideal model organism, because we know the histories and ecologies of different populations. Since the development of agriculture, we’ve had several ongoing natural selection experiments in our species.
Nor can we ignore the longer prehistory of human populations. I tend to think that a lot of recent selection has involved new genetic solutions in cases of strong stabilizing selection. A trait like brain size does not evolve under classic directional selection, but instead as a consequence of shifting patterns of stabilizing selection. With intense selection on multiple functions, such traits are constrained in their evolutionary response. Slight frequency changes are not likely to relax such constraints, but a new mutation of large effect might break a long-standing genetic logjam.
So I think Pritchard and Di Rienzo have outlined many important issues in this review. They have the potential to be highly productive for people with a little talent for applying theory to the data.
References
Pritchard, J. K., & Di Rienzo, A. (2010). Adaptation–not by sweeps alone. Nature Reviews Genetics, 11(10), 665-667. https://doi.org/10.1038/nrg2880
Park, J. H., Wacholder, S., Gail, M. H., Peters, U., Jacobs, K. B., Chanock, S. J., & Chatterjee, N. (2010). Estimation of effect size distribution from genome-wide association studies and implications for future discoveries. Nature genetics, 42(7), 570-575. https://doi.org/10.1038/ng.610

John Hawks Newsletter
Join the newsletter to receive the latest updates in your inbox.

{kind=link}