Top 10 discoveries about ancient people from DNA in 2023
This year's highlights include ways of finding ancient relatives, how some phenotypes evolved in ancient people, and trace evidence from artifacts.
If you're eagerly following discoveries from genetics, this year might have seemed a bit more sedated than last year. After all, 2022 saw a Nobel Prize awarded for innovation in ancient DNA, researchers uncovered a Neandertal family from mere bone fragments, and a team carried out a major study of ancient genomes from eastern Africa.
This year saw a lot of research consolidation, with continued progress along well-established lines. This year saw many research studies that are in this category of “filling in the map” of ancient DNA, especially within the last 3000 years. These included ancient DNA genotyping from Scandinavia, Tibet, the east coast of Africa, northwest Africa, the Aegean, and many more. In a preprint released this year, Swapan Mallick and collaborators have described the Allen Ancient DNA Resource, a database that now includes more than 10,000 individuals.
Building out the database of ancient DNA is undeniably valuable. But even more fascinating are the discoveries that allow us to look at biology, social dynamics, and evolution in new ways.
Discoveries like that don't always get big headlines. This year I read many new studies that deserve a lot more attention than they got when they were published. These include studies of patterns of natural selection in past humans, how humans affected other ancient species, and how different populations interacted with each other. New techniques have opened up avenues of understanding kinship from ancient populations and the links between people and artifacts.
Ancient balancing selection
In January, Alber Aqil and coworkers reported that humans today carry a large number of deletion polymorphisms that persisted in our ancestral populations for more than 700,000 years. The study considered parts of the genome where some people have deletions of more than 50 base pairs compared to sequences found in other people. The research shows that many of these deletion polymorphisms are older than the common ancestral population of recent people and Neandertals. Their age suggests strongly that many of these large deletion polymorphisms were maintained by balancing selection.
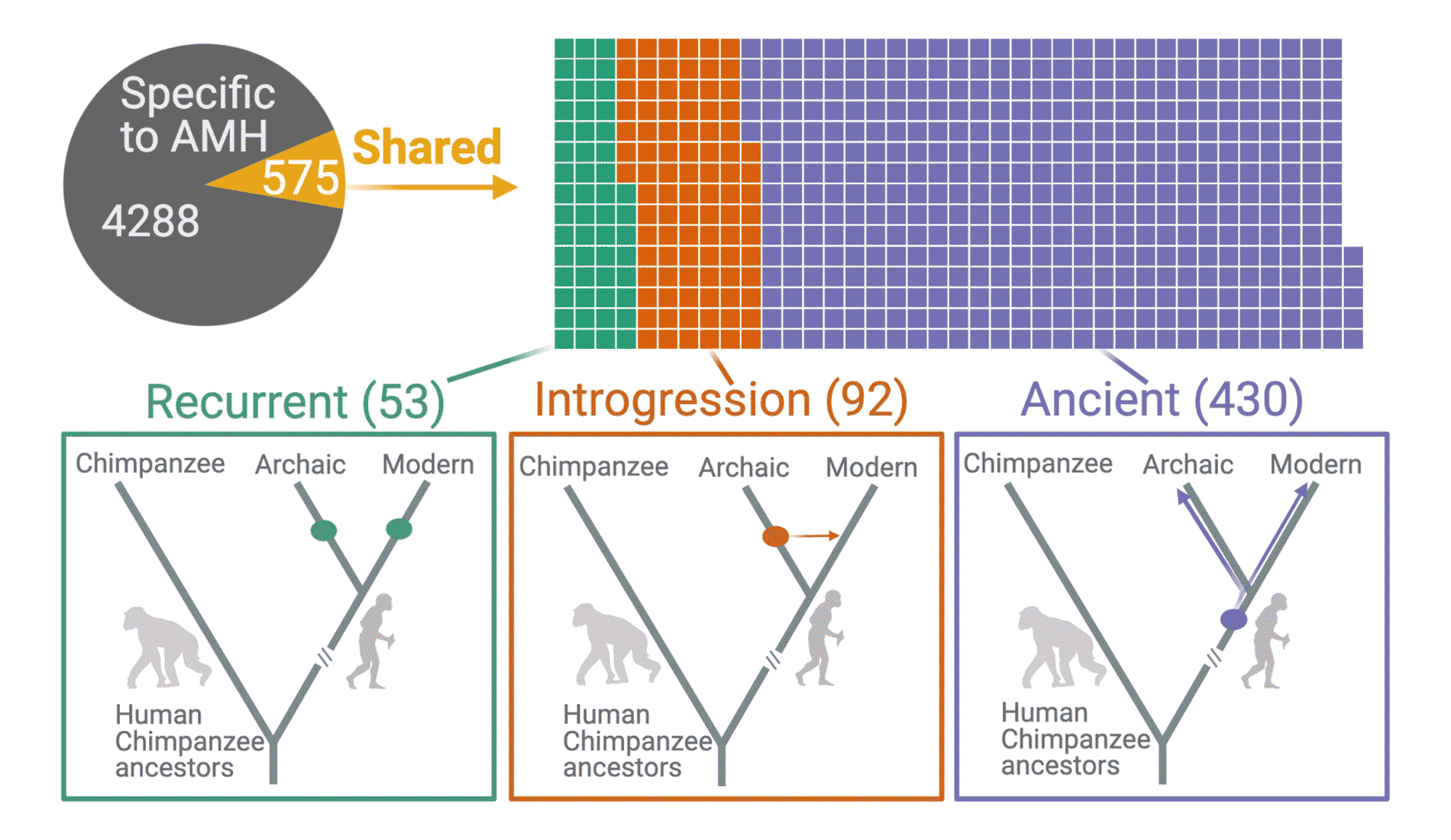
The statistics supporting this observation are very clear. Around 12 percent of large deletion polymorphisms are found in the handful of Neandertal genomes that exist today, and the vast majority of those were retained by both modern and Neandertal populations from their common ancestors. That implies a much higher persistence of these polymorphisms compared to genetic drift alone. Aqil and collaborators showed that many of the deletions intersect with the coding regions of genes, and that these locations show more associations with phenotypes than the average across the genome.
Balancing selection can be a result of overdominance, in which heterozygotes have better survival or reproduction. Or it can result from frequency dependence, where the relative fitness of an allele declines when the allele becomes very common, or balancing selection can happen when an allele has varied fitnesses in different times and places. Aqil and collaborators looked across today's populations to try to assess which of these patterns might have happened more often. What they found was that variation of fitness at different times and places was probably a common pattern.
What does all this mean? For one thing, many of the big missing pieces of genome evidently were useful in some way for ancient survival. If it's true that many polymorphisms reflect balances between fitness effects at different times and places, their persistence in today's populations shows the interaction of ancient groups from different places.
Finding ancient relatives
Many fascinating questions about ancient people involve their social dynamics and kinship. While ancient DNA has given some remarkable insights into the relationships of populations, these finer-scale anthropological questions can be more challenging because they require large local samples and higher coverage datasets. This year, Harald Ringbauer and collaborators from David Reich's lab described a new statistical method for identifying genetic kin from ancient DNA samples. Applied to the growing dataset of ancient DNA sequences from Eurasia, Ringbauer and coworkers characterized dozens of genetic relatives.
Finding long shared single nucleotide polymorphism (SNP) haplotypes is how commercial genotyping approaches like 23andMe match customers to their relatives. Individuals who share a genetic ancestor within three or four generations will share long segments of identical DNA sequence, known as “identity-by-descent” (IBD) blocks. Siblings share half their genomes, on average, second-degree relatives like uncles and nephews share a fourth, and full first cousins share an eighth. Over generations, recombination breaks up these IBD blocks into shorter and shorter lengths, and after six or seven generations it becomes very likely that people don't share any at all.
The problem with ancient genomes is missing data, which poses a challenge to algorithms that rely on matching genotypes. Ringbauer and colleagues tuned their “ancIBD” approach to work well with low-coverage ancient datasets, and as a result the method identifies genetic relatives reliably. This isn't the first statistical tool for identifying ancient relatives, but the ancIBD method is well-matched to the largest samples of ancient genome data.
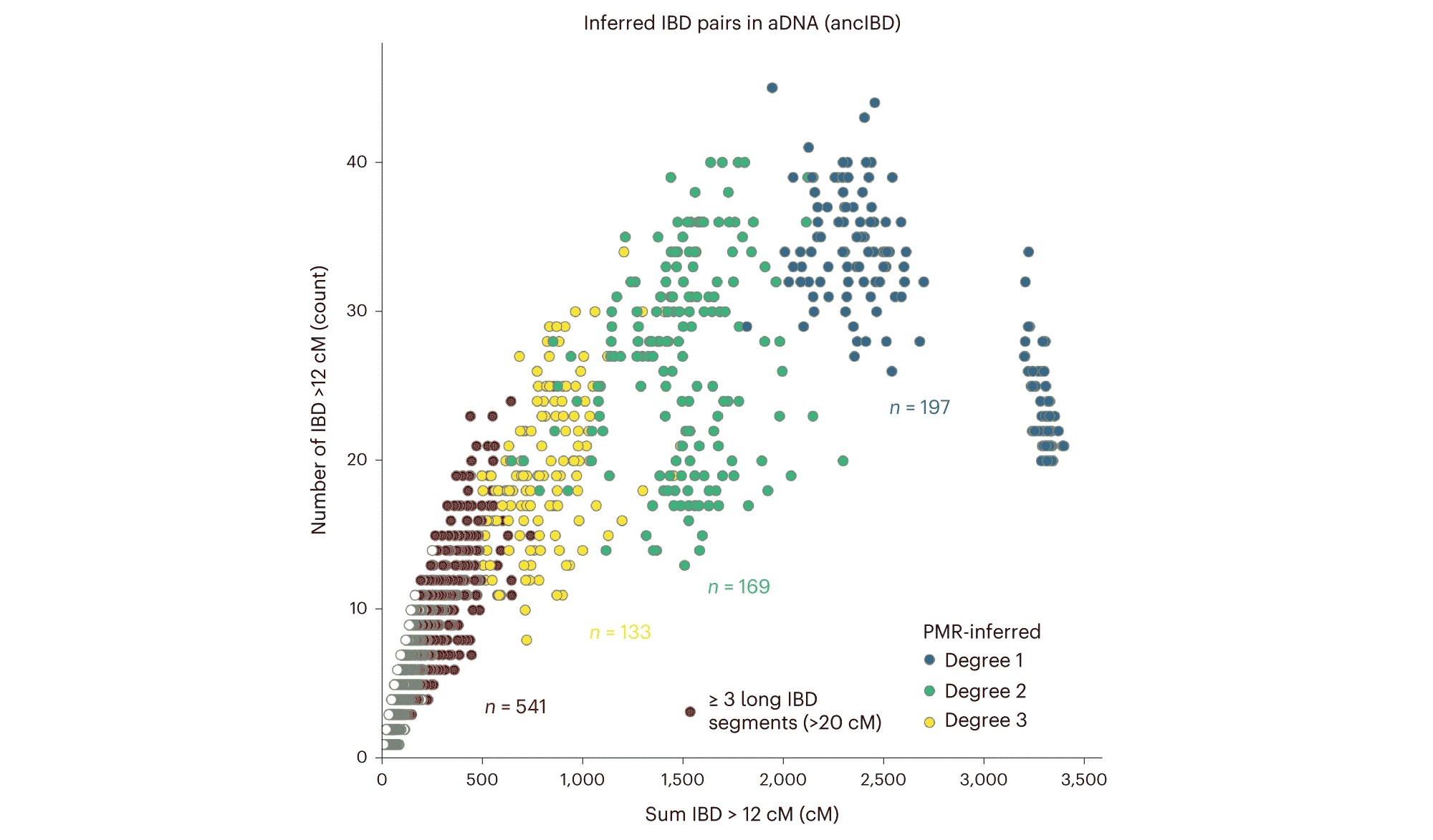
The researchers highlight a few cases in their study. These include genetic relatives among steppe populations of the early Bronze Age, including those associated with the Yamnaya culture. This group has been of great interest to ancient DNA specialists as a possible source population for the Indo-European language family. Ringbauer and coworkers find some relatives of Yamnaya people in burials of the Afanasievo culture of central Asia, representing the same period of time. This evidence of long-distance kin connections helps to document the high migration across the steppe region at the time that early Bronze Age cultures developed.
Smelling like a Denisovan
When Neandertal and Denisovan ancient genomes were first made available for study, I got very interested in olfactory receptor genes as a possible way of understanding how these ancient people and recent people may have adapted to different environments. It takes hundreds of olfactory receptor genes to make our sense of smell. These genes share a common structure and pattern of expression within olfactory neurons but sequence differences cause the genes to bind different molecules that enter the nasal airways. A person's smell universe may differ from other people just because they carry different alleles for these genes. Both natural selection and genetic drift have shaped the landscape of smell in different species. Smell helps to detect tasty foods and potentially poisonous ones, helps track prey animals and find mates.
But olfactory receptors are challenging to study. Mostly it is not possible to predict what a person can smell from odorant receptor genotypes, although researchers have made some progress in matching genes to molecules. With ancient genomes, it's hard to be confident about sequence variations and possible nonfunctional alleles.
This year Claire de March and coworkers examined a sample of 30 olfactory receptor genes in ancient Neandertal and Denisovan genomes. That's in the neighborhood of one tenth of the total number of olfactory receptor genes in the human genome, so it may reflect only a small fraction of the overall repertoire. Recent people have much more variation of these olfactory receptor genes than any of the Neandertal or Denisovan ancient genomes. This accords with the lower genome-wide variation found in these ancient people.
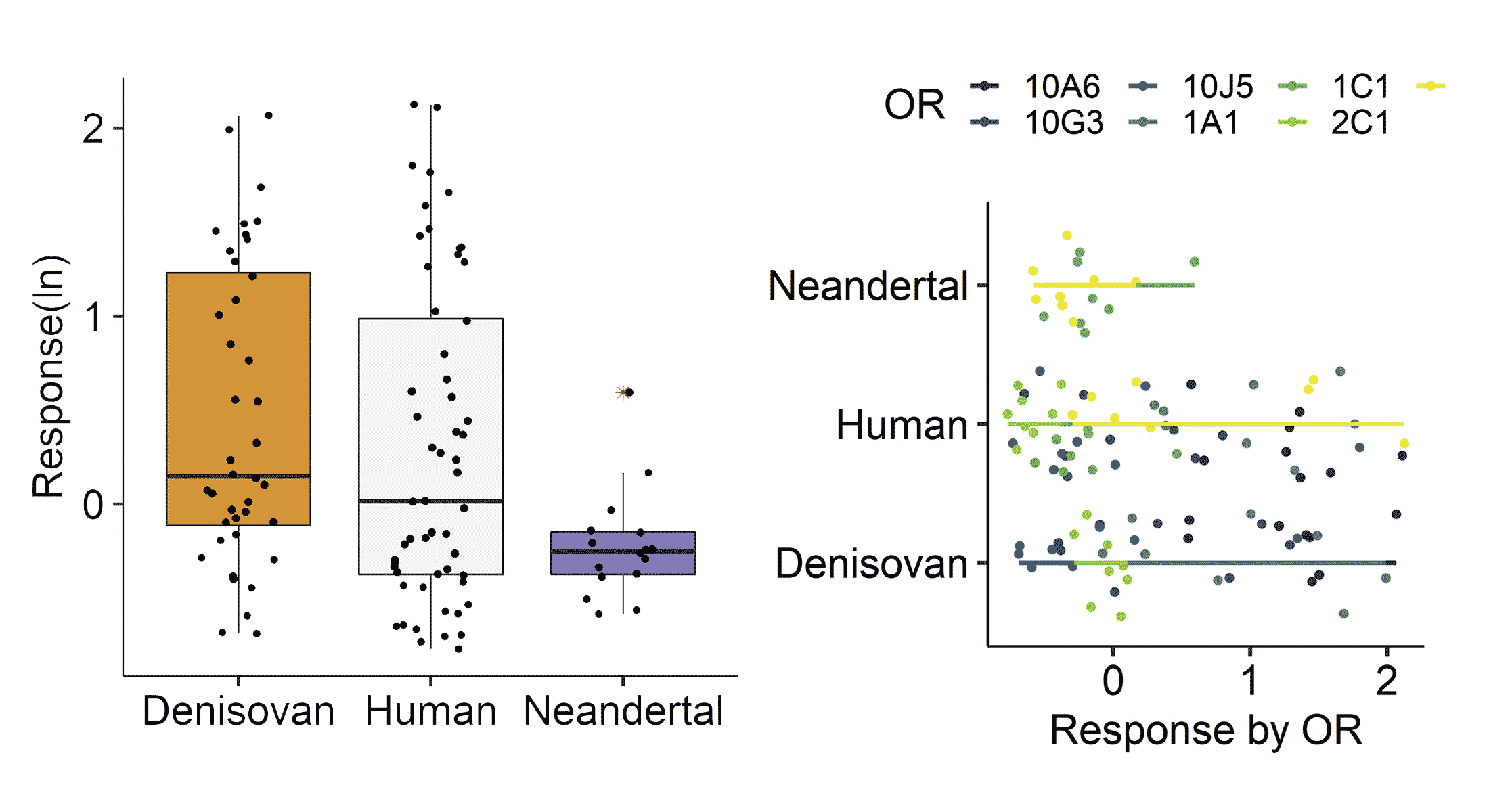
Both Neandertals and Denisovans had a few missense variants in the 30 genes that had not been seen previously in any humans. The researchers tested these amino-acid-changing variants to see their responses to potential odor molecules. What they found was that Denisovan missense variants had a stronger response than their human orthologs, which de March and coworkers narrowed down to “odors contemporary humans perceive as spicy, balsamic, and unpleasant”. Neandertals, on the other hand, had a much lower response for the missense variants which were found in these genes. That result might align with previous research which has shown that Neandertals generally have a higher number of nonfunctional or deleterious alleles across their genomes.
These results provide only hints about the overall scent landscape of these two ancient groups. It's too soon to draw conclusions about adaptations of ancient people from their olfactory receptors. But the study shows what may be gained by biochemical testing of protein variants that are encoded in ancient DNA. If we are going to understand much about the biology of Neandertals from their ancient genomes, that understanding will come from studying the effects and interactions of dozens or hundreds of genes. Olfaction is one biological system where this kind of research may advance.
Selected for immunity
Viruses, bacteria, and parasites hurt and killed a high proportion of people throughout our evolutionary history. The ability of human immune systems to fend off the relentless attacks of these pathogens is a result of natural selection in ancestral human populations. Past research has highlighted a few strong gene-pathogen associations—beta globin variants as a risk factor for malaria, for example. But aside from such strong genetic associations immunity poses a challenge of complexity. Hundreds of genes may be involved in the body's response to any given pathogen. When strong selection is distributed across many genes, it changes most allele frequencies by only a small amount.
Ancient DNA samples in western Eurasia have recently become large enough to detect small changes in allele frequency. A few recent studies have assessed whether alleles that are associated with particular phenotypes changed over time in ancient Europeans. I covered one such study in last year's review.
This year, a study led by Gaspard Kerner looked at genes related to immune response and autoimmune disease. They found that human susceptibility to a broad range of pathogens was under selection during the last 10,000 years, with selection strongest in the Bronze Age and thereafter.
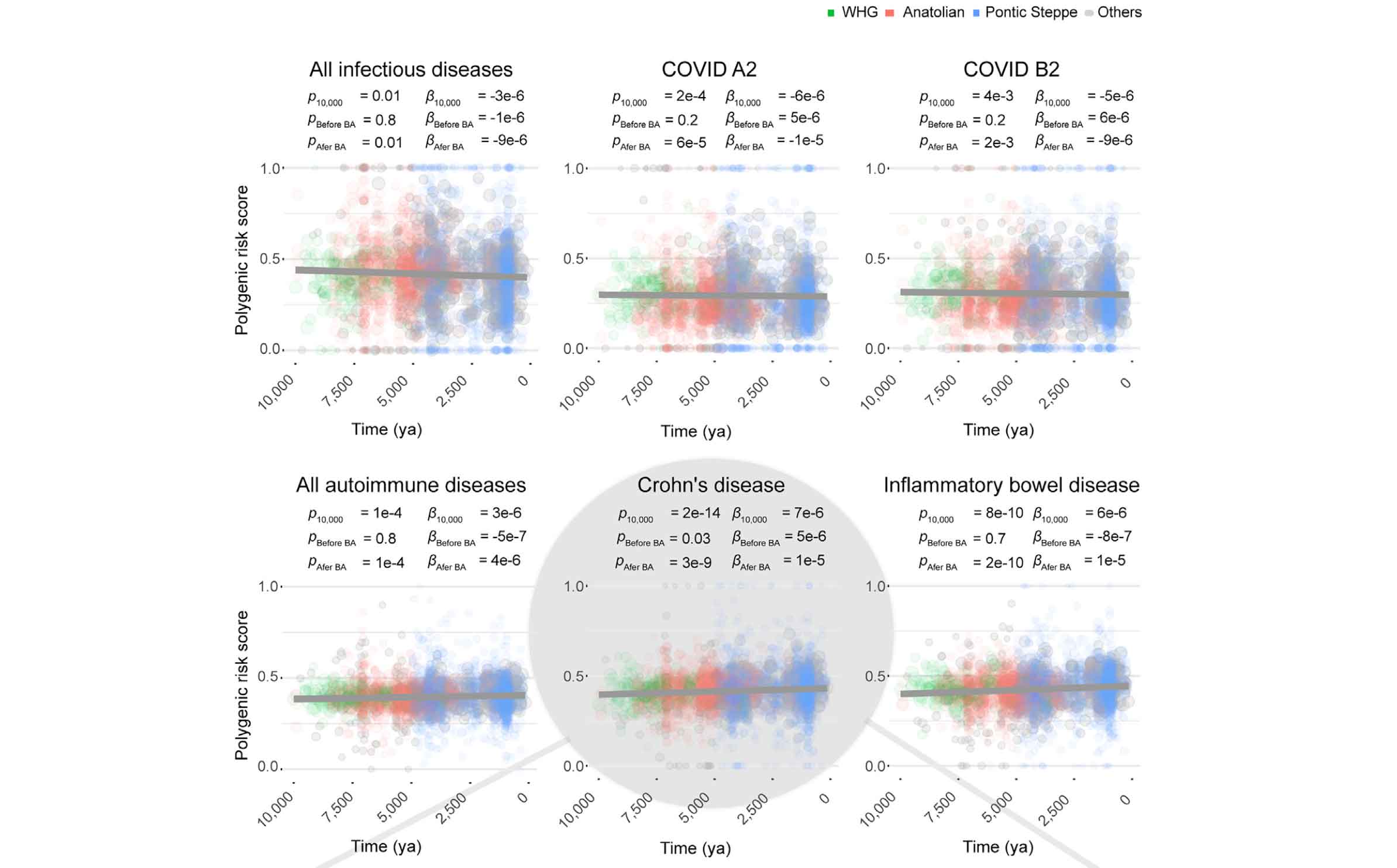
What drew my attention to this study was its estimation of selection coefficients for thousands of alleles across the genome. Similar to some of my own research from fifteen years ago, Kerner and coworkers applied a model that includes the growth of the ancient population, and they examined genetic outliers—in their case, alleles that had a higher estimated selection coefficient than 99% of neutral variants as simulated under their population model. Their dataset enables them to examine time series of allele frequencies directly, making it clear when and how much the frequencies changed. Among the alleles with the strongest estimated selection coefficients, they found a large number that are associated with immunity phenotypes. The strongest allele frequency changes were within the last 5000 years.
An interesting outcome of the study is that selection on immunity had two kinds of outcomes. Resistance to infectious diseases increased over time. Genetic risk factors for autoimmune diseases also increased over time. Kerner and coworkers consider the hypothesis that the appearance of new pathogens may have selected for alleles that have the side effect of increasing autoimmune disease.
Killing the megafauna
Anthropologists tend to focus on DNA from ancient humans, but DNA evidence from other animals and plants is making up a bigger and bigger part of our understanding of human prehistory. One of the biggest long-term scientific questions about ancient people is how much of an impact they had on their environments. No observation is more provocative then the Pleistocene extinctions of megafauna: What role did humans play?
Going back to the nineteenth century, naturalists thought that the Ice Age megafauna became extinct when the climate changed to the much warmer conditions of the present day. During the 1960s, the paleontologist Paul Martin proposed an alternative: Ancient humans had hunted the prehistoric beasts to extinction. Martin built this “Pleistocene overkill hypothesis” from the observation extinctions across different continents and islands did not correspond with the global end of the last glaciation. In some regions, especially the Americas, Australia, and Madagascar, extinctions seemed to follow the first arrival of humans. In defense of the climate-induced extinction hypothesis, many researchers have focused upon lesser-known climate events that preceded the Last Glacial Maximum but may nonetheless have made survival harder for megafauna.
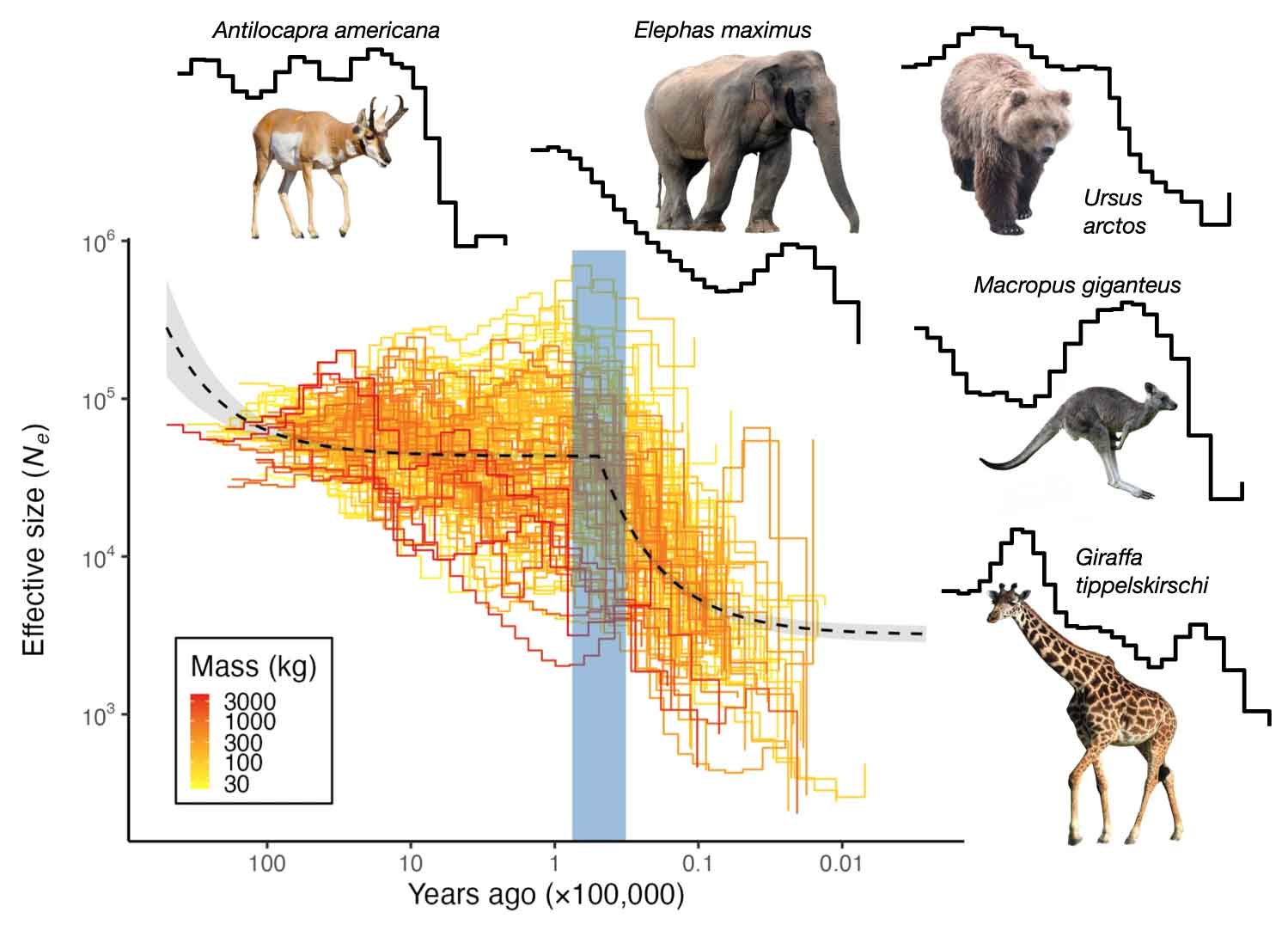
Not only extinct species but also surviving large mammals suffered population declines during the Late Pleistocene. This year, Juraj Bergman and collaborators used DNA from more than 100 species of Pleistocene survivors to examine when each species declined in numbers. At the moment this is a much larger sample of genetic information than has been obtained from ancient DNA of extinct species. It encompasses not only the largest surviving megafauna like elephants and rhinoceros, but also medium-sized mammals. These species show substantial disruptions in their demographic histories during the Late Pleistocene. Bergman and coworkers found a shared pattern: a collapse in numbers between 76,000 and 32,000 years ago. That timeframe is earlier than the Last Glacial Maximum around 18,000 years ago, and also much earlier than the end of the Pleistocene 10,000 years ago.
The timeline of known climate events does not match up well with these demographic reconstructions. The species did not share any consistent pattern across previous glacial cycles, giving weight to the often-heard argument that so many species survived every Pleistocene glaciation but the last one. Bergman and coworkers found that the shared pattern of collapses could be explained by considering a model that includes the times of human habitation for regions where each species lives.
The species in this study are still alive, which means they survived the depredations of ancient humans. But some are hanging on only by a thread, threatened or endangered within their native habitats. The megafaunal species that became extinct during the Late Pleistocene and Holocene were for some time just like these threatened survivors. This new research will surely not end the long scientific debate about the causes of megafaunal extinctions. But it provides strong evidence that humans were an important part of the picture.
Climate and populations of pre-Hispanic Mexico
In May, Viridiana Villa-Islas and coworkers reported on the genomes of 12 individuals from northern and central Mexico, dating to between 600 CE and 1400 CE. They also recovered the mitochondrial genomes of an additional 15 people, going as far back as 300 BCE. There has been quite a lot of earlier work on mitochondrial DNA from Mesoamerica, but nuclear genomic data has previously come only from a small number of individuals, and only at very low coverage.
Villa-Islas and collaborators shaped their study to test an idea about climate change that had originated from the archaeological record. The climate record shows that there were prolonged droughts in the region between 900 and 1300 CE, corresponding to the time known as the Medieval Warm Period. Some archaeological evidence from north-central Mexico had suggested that these droughts caused peoples from the arid north to move south, displacing some populations of central Mexico in the process. The genomes included in the study includes some from four sites in the region where replacement was proposed to have happened.

The genome data do not confirm the replacement idea, although they don't fully settle the issue. One of the sites had been abandoned before 1100 CE, so a direct comparison before and after the period of drought for this site was not possible. The other three sites came from the area known as the Sierra Gorda, where genomes showed evidence of continuity and no evidence of replacement. The authors considered it possible that conditions in this area may have been uncharacteristic of the broader region.
The most interesting result of the study was an unexpected one. The DNA did not show evidence for population movement within the time period represented by these genomes, but it did provide hints about much earlier events. Villa-Islas and coworkers identified “ghost population” mixture, finding that two of the samples of ancient genomes have genetic heritage from earlier groups that are not represented by DNA samples. They estimated that one of these ancient groups diverged around 24,500 years ago from the ancestors of other Native American populations. This result points to diversification among the first inhabitants of the Americas, peoples that established new populations and later came back into contact with each other.
Intricacies of the European Upper Paleolithic
Early in the year, Cosimo Posth and coworkers provided a new survey of European population history from the early Upper Paleolithic around 35,000 years ago up to the advent of the Neolithic 5000 years ago. Their study included 116 new ancient genomes, which they combined with earlier data for a total of 356 individuals. This sample gave the researchers the ability to look for shared patterns of ancestry in different parts of Europe during several cultural periods, from Gravettian cultures more than 25,000 years ago, to Solutrean, Epigravettian, and other cultures around the Last Glacial Maximum, up to Final Paleolithic and Mesolithic cultures across the continent.
Posth and coworkers confirmed that the people that archaeologists have recognized as the Gravettian culture were no one population. Instead, “Gravettian” is a category including at least two populations with different ancesties in eastern and southwest Europe. This finding implies a high degree of cultural exchange between groups across Europe with different population histories. Another paper this year from Vanessa Villalba-Mouco and collaborators came to a similar conclusion, using the genome of a Solutrean-associated individual from Iberia as leverage on the question.
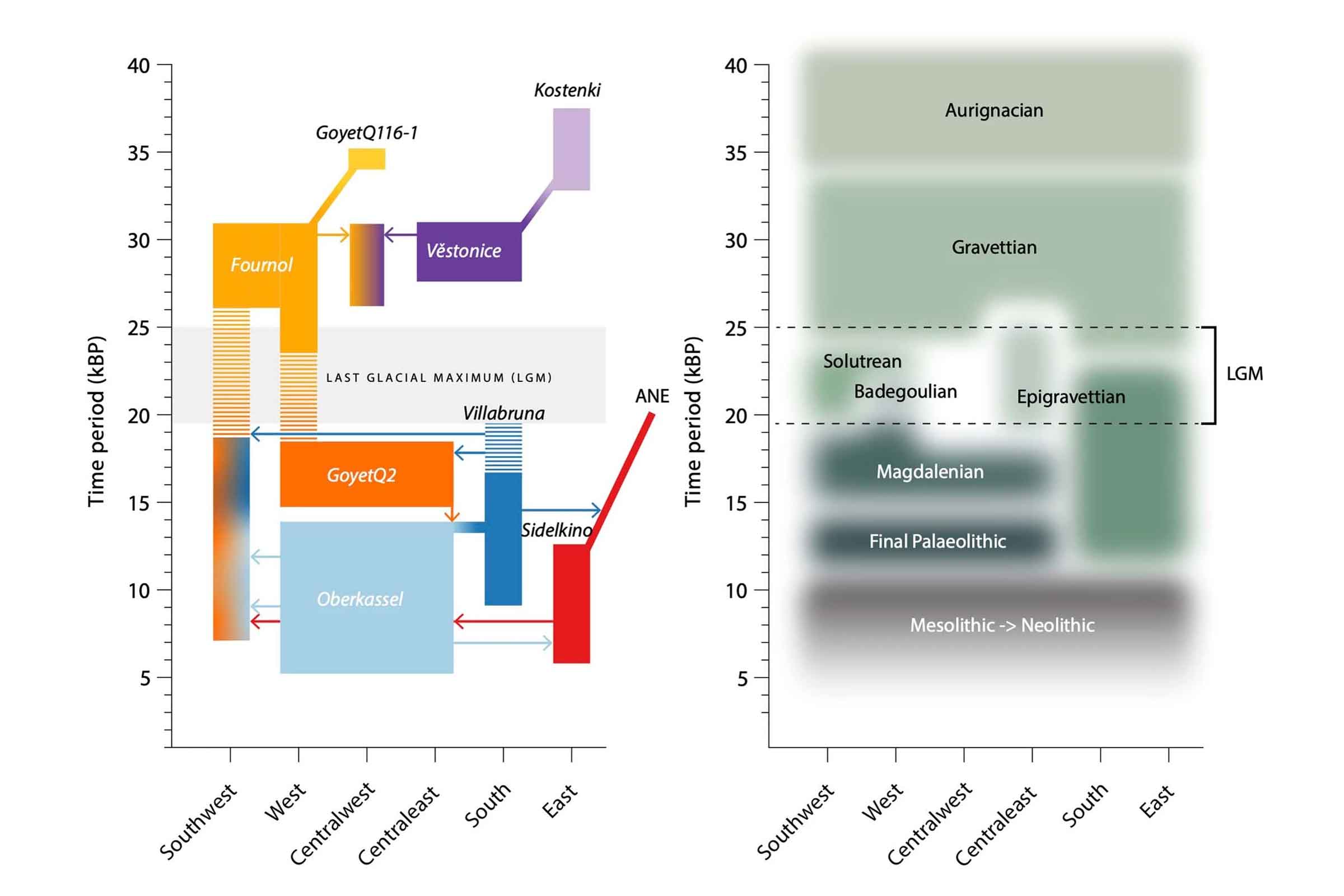
Stepping forward in time, archaeologists have long known that northern parts of Europe were depopulated and recolonized from Iberia, Italy, and the Balkans as the ice receded. The new study adds some detail to these movements. Genomes from Italy and nearby areas after the Last Glacial Maximum confirm that many ancient people were related to a Late Paleolithic individual from Ripari Villabruna in northern Italy. This “Villabruna cluster” of ancient people had no close connection to people in the area before the Last Glacial Maximum, and here the researchers suggest this group may have originated in the Balkans. The Magdalenian culture, widespread across western and central Europe, was made up of at least two populations that had different ancestry and mixture with these Villabruna cluster people. Finally, by the end of the Pleistocene a group connected with central Asia appears in eastern Europe. This put into place a population structure that would interact with Neolithic populations.
As big as this ancient DNA sample is, it adds up to a little more than one individual per century across the whole continent. That leaves much more to discover. What's clear that the cultures identified by archaeologists are different entities from populations as conceived by geneticists. Transitions of cultures often marked the arrival of new people, but dispersion of a culture seems often to have happened across populations with diverse ancestries.
A source for the braided stream
Two related papers this year examined the deep population history of Africa. One of them, by Aaron Ragsdale and coworkers, traced ancient gene flow and population structure back to around a million years ago. The other, by Wangjie Hu and colleagues, proposed that a tight bottleneck occurred in African ancestral people around that same time. Together they sketch out a history in which the deep relationships of today's people were an interlinked network going back to the Early Pleistocene.
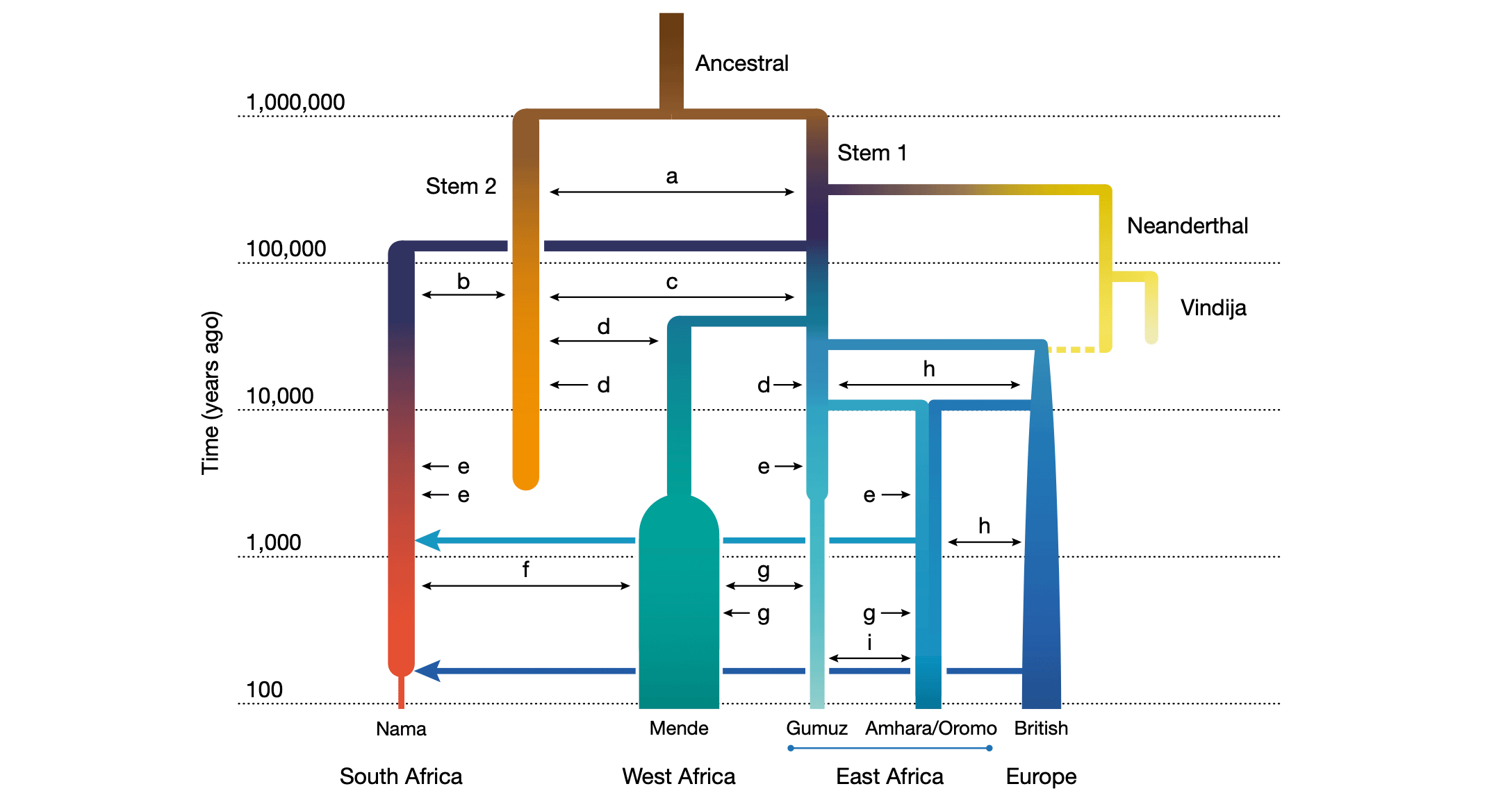
This scenario has some important implications. For one thing, the interconnections of African populations have a deeper history than the emergence of Neandertal and Denisovan ancestors into Eurasia. Whatever started ancient humans on the path toward today's populations, these ancient groups were all part of the same story. Another implication: A bottleneck and diversification of humans around a million years ago was long after the first differentiation of H. erectus and its appearance across Asia. Whether so-called superarchaic gene flow traced within some Denisovan and Neandertal genomes might have come from the more ancient H. erectus diversification is not yet clear.
This research will not be the last word. The growth of datasets has enabled them to test more intricate models than the simple trees of a decade ago. But where ancient DNA has been available in Eurasia, it has generally found unexpected groups. There were surely many of those in our African heritage.
Trace evidence from ancient jewelry
Bones and teeth are no longer the only sources of ancient genetic evidence. For a few years, researchers have been pulling DNA from sediment. I covered some fundamental work on sediment DNA as one of the advances of 2022. In some northern European contexts from the last 10,000 years, DNA has also been sequenced from birch pitch or gum that was chewed by ancient people. This year, Elena Essel and coworkers from Matthias Meyer's research group found human DNA on the surfaces ancient pendants, up to 45,000 years old.
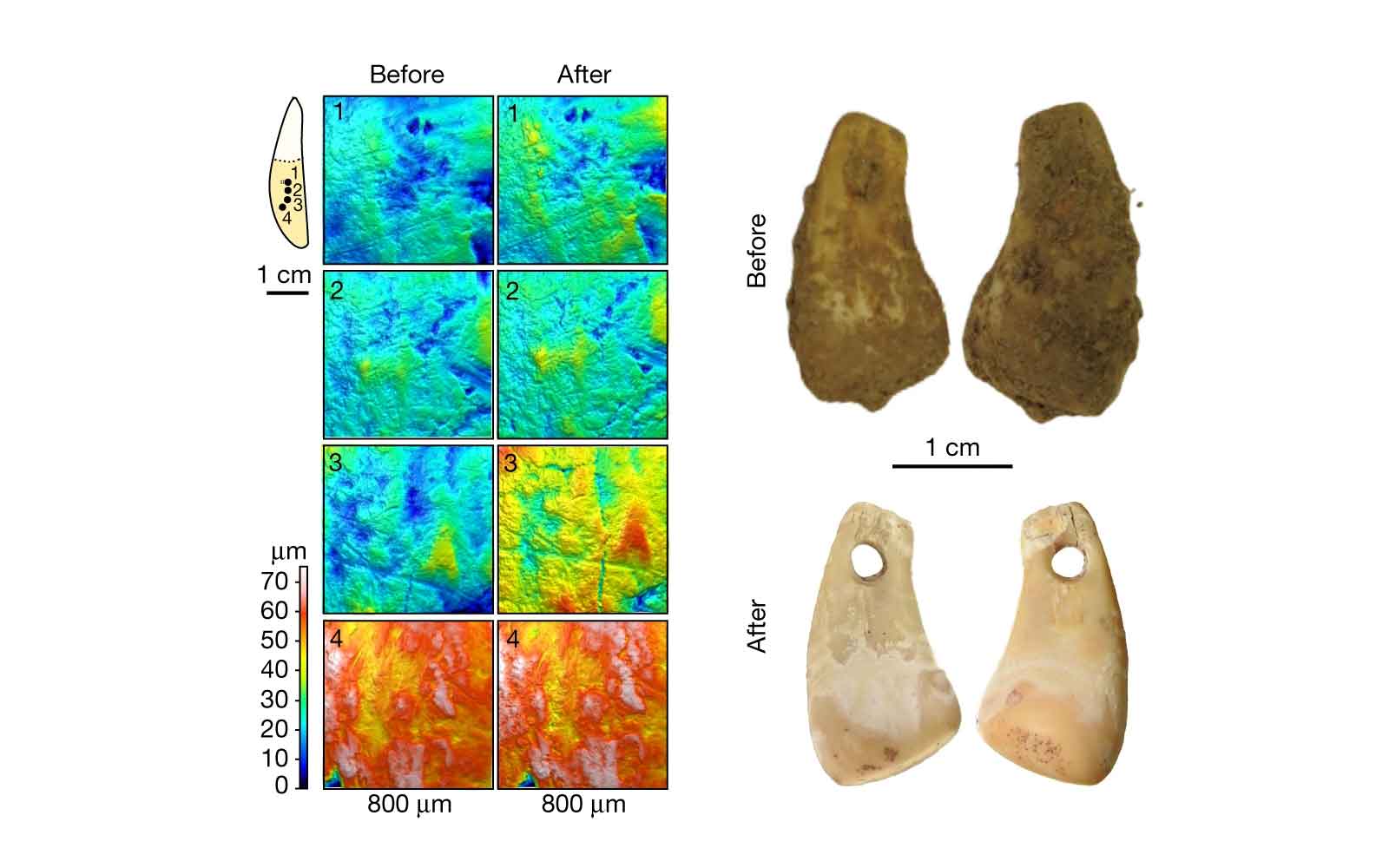
The researchers developed an original method for sampling DNA from artifacts without drilling. The method involves a series of steps in which the artifacts are soaked in a solution at different temperatures, giving up some absorbed DNA molecules into the solution. For a cervid tooth pendant from Denisova Cave, this process yielded both the deer's DNA and a human mitochondrial sequence. Their investigation of the pendant also gave them enough human nuclear DNA to identify the ancient person's female genetic sex, as well as placing the individual near other ancient Siberian people in a principal components plot.
The study was a proof of principle in which the Denisova pendant gave the best results. Ten artifacts from two French sites, which had come from past excavations with normal archaeological cleaning and handling, did not generate results that were as promising. In their conclusions, Essel and colleagues stressed that their approach worked the best with freshly excavated artifacts that had not been handled directly by archaeologists. They note the possibility of working out the social dimensions of artifact manufacture and use, including gendered use of artifacts, if the method could be applied at a large scale.
Indigenous Australian population history
In December, two studies reported results from a new project to sequence genomes of Indigenous Australian people. The work was conducted under a collaboration by the National Centre for Indigenous Genomics, a research center established to enable Indigenous custodianship over biological samples at the Australian National University. Under the collaboration, 159 people representing several Indigenous communities participated in whole-genome sequencing. The two research studies present the first results on their variation and population structure, and the occurrence of structural genome variants in the sample.
Australia and Papua New Guinea together were a single landmass until after 12,000 years ago, known by geographers as Sahul. From the first habitation of the continent before 50,000 years ago up to the rise of the sea level, populations across this region could have been connected. The genetic data show that the separation of Papuan and Australian groups emerged by 30,000 years ago. The ancestors of the Indigenous Australian groups who participated in the study were themselves beginning to separate by 19,000 years ago.
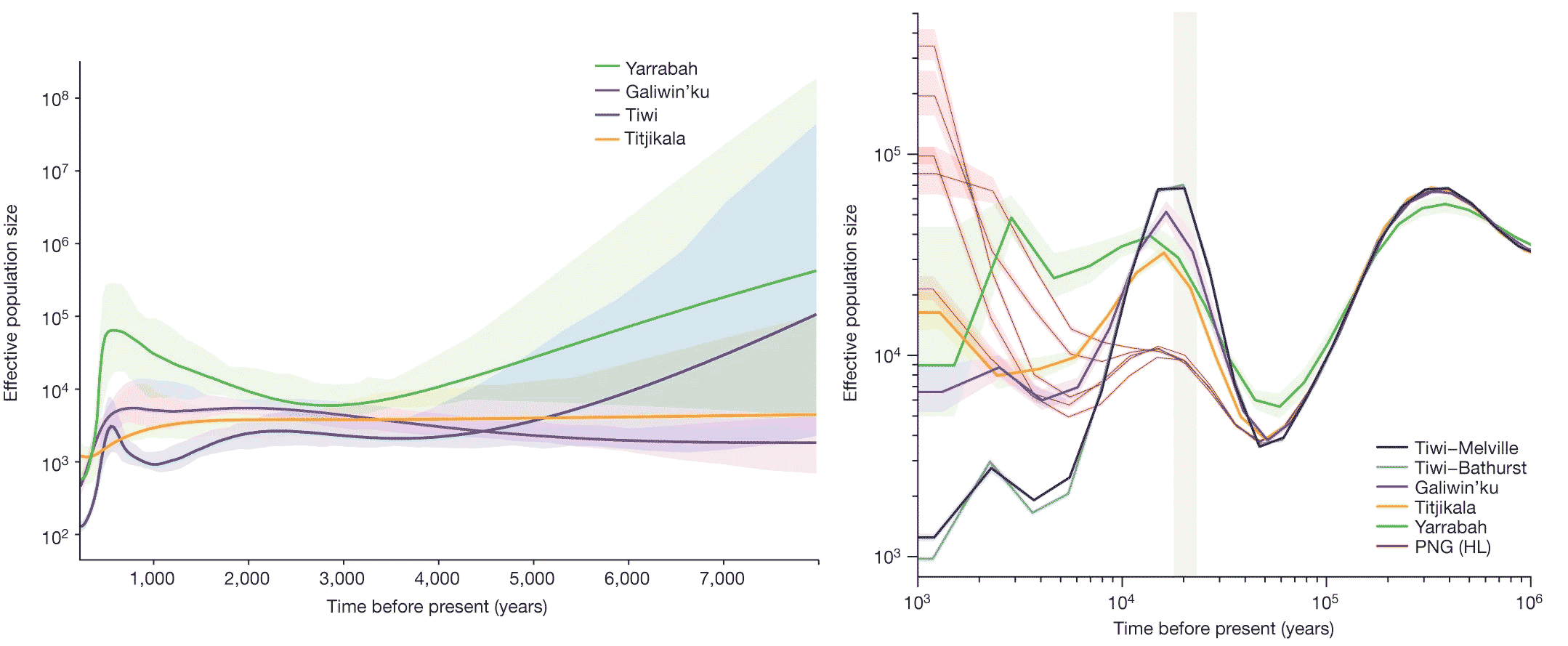
The communities who were part of the study mostly include speakers of languages in the Pama-Nyungan language family, the most widespread linguistic grouping across Australia. The exceptions are three communities from the Tiwi islands, who speak the Tiwi language. These groups have a distinctive history with a long separation from mainland Australian populations. While the genetic variation in all these groups is low, their variation is distinctive in comparison with other regions of the world. As a corollary, the study of structural variants by Andre Reis and coworkers found that these groups had more unique variants than individuals from other populations.
Such observations may open valuable insights for health of Indigenous Australians. One of the most important ways this study differs from earlier research is that Indigenous people helped shape the research design from the start. When geneticists collaborate with communities on research questions that people value, it builds a stronger foundation for discovery.
Deeper reading
There is much more to learn by continuing to develop broader and deeper samples of individuals from all parts of the world. A growing proportion of that research is being done with the leadership of researchers from the countries where ancient remains are found, with the involvement of communities. In some cases, descendants of ancient people are now collaborators with research. In April, a review by Emma Kowal and twenty coauthors stressed the ethical importance of involving communities in the planning and processes of research.
Many researchers this year have reflected on ways that practices in ancient genetics are changing. In an October special issue of Science, María Ávila-Arcos, Maanasa Raghavan, and Carina Schlebusch focused on some of the achievements of paleogenomics as well as the challenges moving forward with community involvement. Their review is a great way for readers to see the current landscape of research and how it fits into our understanding of population history.
Meanwhile, the anthropologist David Anthony this June published an essay that ponders some of the misunderstandings between archaeologists and geneticists about migrations. From my perspective, his essay raises the simple question of whether archaeologists and geneticists have been studying the same thing. Genetic data can reveal mating networks but are silent about the causes and consequences of movement; archaeological data pertains to ecology and culture, which may say little about the mating or movement of people. Anthony's last words are a message for the future, as he notes that researchers are “only beginning to build the tools necessary to understand the social, linguistic, and economic effects of migrations”.
Each of these three reviews gives a perspective on how anthropological science is adapting to integrate discoveries from genetics. DNA data itself has answered some valuable questions, setting our understanding of human evolution and prehistory onto a new path. What lies before the field is finding ways to explore the functional, social, and ecological networks that shaped DNA variation in the past.
Notes: I wrote at some length this year about ancient gene flow within Africa and the paper by Hu and coworkers that describes an ancient bottleneck. The papers come to somewhat different conclusions but are based on similar datasets. Comparing their results shows that what may seem like differences in their numbers are a consequence of assuming different mutation rates.
The previous work on ancient human DNA from birch mastics is well worth reading, including the 2019 work by Natalija Kashuba and coworkers, which investigated eight preserved mastics from a Mesolithic site in Sweden.
Interested readers can find my 2022 list of top discoveries, which covers a few different areas of research than this year's list.
References
Anthony, D. W. (2023). Ancient DNA and migrations: New understandings and misunderstandings. Journal of Anthropological Archaeology, 70, 101508. https://doi.org/10.1016/j.jaa.2023.101508
Aqil, A., Speidel, L., Pavlidis, P., & Gokcumen, O. (2023). Balancing selection on genomic deletion polymorphisms in humans. eLife, 12, e79111. https://doi.org/10.7554/eLife.79111
Ávila-Arcos, M. C., Raghavan, M., & Schlebusch, C. (2023). Going local with ancient DNA: A review of human histories from regional perspectives. Science, 382(6666), 53–58. https://doi.org/10.1126/science.adh8140
Bergman, J., Pedersen, R. Ø., Lundgren, E. J., Lemoine, R. T., Monsarrat, S., Pearce, E. A., Schierup, M. H., & Svenning, J.-C. (2023). Worldwide Late Pleistocene and Early Holocene population declines in extant megafauna are associated with Homo sapiens expansion rather than climate change. Nature Communications, 14(1), Article 1. https://doi.org/10.1038/s41467-023-43426-5
Essel, E., Zavala, E. I., Schulz-Kornas, E., Kozlikin, M. B., Fewlass, H., Vernot, B., Shunkov, M. V., Derevianko, A. P., Douka, K., Barnes, I., Soulier, M.-C., Schmidt, A., Szymanski, M., Tsanova, T., Sirakov, N., Endarova, E., McPherron, S. P., Hublin, J.-J., Kelso, J., … Meyer, M. (2023). Ancient human DNA recovered from a Palaeolithic pendant. Nature, 618(7964), Article 7964. https://doi.org/10.1038/s41586-023-06035-2
Hu, Wangjie, Hao, Ziqian, Du, Pengyuan, Di Vincenzo, Fabio, Manzi, Giorgio, Cui, Jialong, Fu, Yun-Xin, Pan, Yi-Hsuan, & Li, Haipeng. (2023). Genomic inference of a severe human bottleneck during the Early to Middle Pleistocene transition. Science, 381(6661), 979–984. https://doi.org/10.1126/science.abq7487
Kashuba, N., Kırdök, E., Damlien, H., Manninen, M. A., Nordqvist, B., Persson, P., & Götherström, A. (2019). Ancient DNA from mastics solidifies connection between material culture and genetics of Mesolithic hunter–gatherers in Scandinavia. Communications Biology, 2(1), Article 1. https://doi.org/10.1038/s42003-019-0399-1
Kerner, G., Neehus, A.-L., Philippot, Q., Bohlen, J., Rinchai, D., Kerrouche, N., Puel, A., Zhang, S.-Y., Boisson-Dupuis, S., Abel, L., Casanova, J.-L., Patin, E., Laval, G., & Quintana-Murci, L. (2023). Genetic adaptation to pathogens and increased risk of inflammatory disorders in post-Neolithic Europe. Cell Genomics, 3(2), 100248. https://doi.org/10.1016/j.xgen.2022.100248
Kowal, E., Weyrich, L. S., Argüelles, J. M., Bader, A. C., Colwell, C., Cortez, A. D., Davis, J. L., Figueiro, G., Fox, K., Malhi, R. S., Matisoo-Smith, E., Nayak, A., Nelson, E. A., Nicholas, G., Nieves-Colón, M. A., Russell, L., Ulm, S., Vergara-Silva, F., Villanea, F. A., … Tsosie, K. S. (2023). Community partnerships are fundamental to ethical ancient DNA research. Human Genetics and Genomics Advances, 4(2), 100161. https://doi.org/10.1016/j.xhgg.2022.100161
Mallick, S., Micco, A., Mah, M., Ringbauer, H., Lazaridis, I., Olalde, I., Patterson, N., & Reich, D. (2023). The Allen Ancient DNA Resource (AADR): A curated compendium of ancient human genomes. bioRxiv, 2023.04.06.535797. https://doi.org/10.1101/2023.04.06.535797
March, C. A. de, Matsunami, H., Abe, M., Cobb, M., & Hoover, K. C. (2023). Genetic and functional odorant receptor variation in the Homo lineage. iScience, 26(1). https://doi.org/10.1016/j.isci.2022.105908
Martin, P. S. (1966). Africa and Pleistocene Overkill. Nature, 212(5060), Article 5060. https://doi.org/10.1038/212339a0
Posth, C., Yu, H., Ghalichi, A., Rougier, H., Crevecoeur, I., Huang, Y., Ringbauer, H., Rohrlach, A. B., Nägele, K., Villalba-Mouco, V., Radzeviciute, R., Ferraz, T., Stoessel, A., Tukhbatova, R., Drucker, D. G., Lari, M., Modi, A., Vai, S., Saupe, T., … Krause, J. (2023). Palaeogenomics of Upper Palaeolithic to Neolithic European hunter-gatherers. Nature, 615(7950), Article 7950. https://doi.org/10.1038/s41586-023-05726-0
Ragsdale, A. P., Weaver, T. D., Atkinson, E. G., Hoal, E. G., Möller, M., Henn, B. M., & Gravel, S. (2023). A weakly structured stem for human origins in Africa. Nature, 1–9. https://doi.org/10.1038/s41586-023-06055-y
Reis, A. L. M., Rapadas, M., Hammond, J. M., Gamaarachchi, H., Stevanovski, I., Ayuputeri Kumaheri, M., Chintalaphani, S. R., Dissanayake, D. S. B., Siggs, O. M., Hewitt, A. W., Llamas, B., Brown, A., Baynam, G., Mann, G. J., McMorran, B. J., Easteal, S., Hermes, A., Jenkins, M. R., Patel, H. R., & Deveson, I. W. (2023). The landscape of genomic structural variation in Indigenous Australians. Nature, 624(7992), Article 7992. https://doi.org/10.1038/s41586-023-06842-7
Ringbauer, H., Huang, Y., Akbari, A., Mallick, S., Olalde, I., Patterson, N., & Reich, D. (2023). Accurate detection of identity-by-descent segments in human ancient DNA. Nature Genetics, 1–9. https://doi.org/10.1038/s41588-023-01582-w
Silcocks, M., Farlow, A., Hermes, A., Tsambos, G., Patel, H. R., Huebner, S., Baynam, G., Jenkins, M. R., Vukcevic, D., Easteal, S., & Leslie, S. (2023). Indigenous Australian genomes show deep structure and rich novel variation. Nature, 624(7992), Article 7992. https://doi.org/10.1038/s41586-023-06831-w
Villa-Islas, V., Izarraras-Gomez, A., Larena, M., Campos, E. M. P., Sandoval-Velasco, M., Rodríguez-Rodríguez, J. E., Bravo-Lopez, M., Moguel, B., Fregel, R., Garfias-Morales, E., Medina Tretmanis, J., Velázquez-Ramírez, D. A., Herrera-Muñóz, A., Sandoval, K., Nieves-Colón, M. A., Zepeda García Moreno, G., Villanea, F. A., Medina, E. F. V., Aguayo-Haro, R., … Ávila-Arcos, M. C. (2023). Demographic history and genetic structure in pre-Hispanic Central Mexico. Science, 380(6645), eadd6142. https://doi.org/10.1126/science.add6142
Villalba-Mouco, V., van de Loosdrecht, M. S., Rohrlach, A. B., Fewlass, H., Talamo, S., Yu, H., Aron, F., Lalueza-Fox, C., Cabello, L., Cantalejo Duarte, P., Ramos-Muñoz, J., Posth, C., Krause, J., Weniger, G.-C., & Haak, W. (2023). A 23,000-year-old southern Iberian individual links human groups that lived in Western Europe before and after the Last Glacial Maximum. Nature Ecology & Evolution, 7(4), Article 4. https://doi.org/10.1038/s41559-023-01987-0

John Hawks Newsletter
Join the newsletter to receive the latest updates in your inbox.



{kind=link}