
When did human chromosome 2 fuse?
More and more, it looks like this event happened shortly before a million years ago, in the common ancestors of Neandertal, Denisovan, and African ancestral humans.

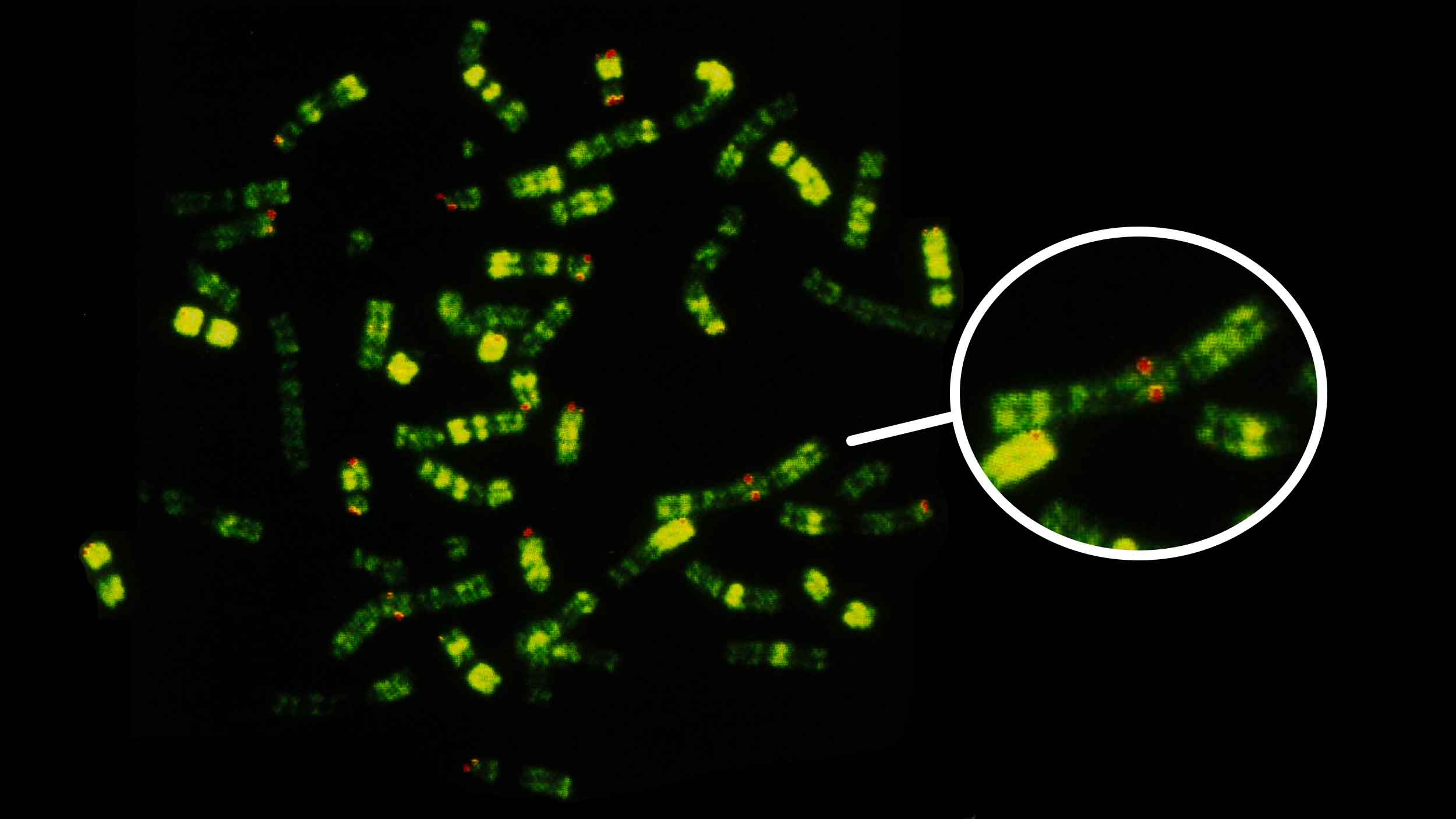
Most living people have 23 pairs of chromosomes, as did our relatives known from ancient DNA, the Neandertals and Denisovans. All of our closest primate relatives—chimpanzees, bonobos, gorillas, and orangutans—have 24 pairs. From comparing these genomes with each other, it is clear that sometime during human evolution two ancestral chromosomes fused together, reducing our number. The product of that fusion is human chromosome 2, the second-largest of our chromosomes.
Evolutionary geneticists often focus closely on differences in chromosome numbers, which can be related to reproductive incompatibility between sister species. Structural changes to chromosomes may also affect gene regulation or other functions. Did the chromosome 2 fusion make a difference to our ancestors, maybe even cause an ancient speciation in our lineage?
Maybe the biggest barrier to understanding how this chromosome fusion mattered is a better knowledge of exactly when it happened. Recently geneticists led by Barbara Poszewiecka applied a creative approach to estimate how long ago the two ancestral chromosomes fused together. They found a surprisingly recent range of times: between 400,000 and 1.5 million years ago.
A new paper just published suggests that a major bottleneck in our evolutionary history happened between 930,000 and 800,000 years ago, and points to the chromosome 2 fusion as one possible consequence. This interval is a very interesting time. Our African ancestors, Neandertal ancestors, and Denisovan ancestors all diverged from each other around 700,000 years ago—and all these branches share the fused chromosome. It seems likely that the population that gave rise to these later hominins was the one in which the chromosome 2 fusion first evolved. That may have made a big difference to their interaction with other hominins that lived at the same time.
Characterizing the fusion
Geneticists demonstrated the correspondences of human and great ape chromosomes more than 40 years ago, visualizing their chromatin content by using special chemical stains. The resulting pattern of bands, still used in genetic maps, makes it very clear which chromosomes are homologous among these species and how they have changed. Aside from our fused chromosome 2, in other ways the human arrangement of chromosomes is actually conservative. For example, the common ancestors of chimpanzees and bonobos underwent pericentric inversions on seven different chromosomes, compared with only two for our ancestral lineage.

Two great ape chromosomes correspond to the two halves of our chromosome 2, and geneticists often designate these as 2a and 2b. The chimpanzee and bonobo chromosomes are a closer match to ours than gorillas, because the human-chimpanzee ancestral population had a pericentric inversion of this ancestral chromosome, reversing the area immediately around its centromere.
When the ancestral chromosomes fused together, the shorter arm of ancestral 2a became connected to the shorter end of ancestral 2b. For some time the newly-fused chromosome had two centromeres. This is an unstable situation. Today the 2a centromere continues to function, while ancestral 2b centromere is just a tiny shadow of its former self. In a 2017 study, Georgia Chiatante and coworkers investigated how this happened. Centromeres differ in sequence and composition, but what they share is a large number of DNA repeats known as α (alpha) satellites, organized into arrangements that form higher-order repeat (HOR) units that are repeated hundreds or thousands of times. These sequence features interact with specialized histones and epigenetic processes to assemble the kinetochore protein complex that enables DNA replication. In human ancestors, a major deletion had eliminated most of the α satellite content of 2b, including most HOR structure.
Still, this nonfunctional centromere provides some of the strongest evidence that the known Neandertal and Denisova 3 genomes share the chromosome 2 fusion. Karen Miga surveyed the ancestral 2b centromere in humans and found several sequence variations in the degraded α satellite repeats that are not found in the chimpanzee genome. The known ancient genomes have these human-specific markers. This is not only evidence that the chromosomes had fused in the common ancestors of ancestral African, Neandertal, and Denisovan populations, but also that the ancestral 2b centromere had already evolved into its humanlike nonfunctional form before these hominins diverged.
Miga additionally noted the presence of a retrotransposon insertion in this ancestral 2b centromere, known as an SVA element. This particular element belongs to a human-specific family known as SVA-E, which began to proliferate in ancestral hominins sometime after our divergence from chimpanzees and bonobos. Hui Wang and collaborators characterized this family in a 2005 paper, estimating that SVA-E began to proliferate after 3.5 million years ago. This date doesn't tell us much about when the centromere became deactivated, however, since the insertion may have happened long after 3.5 million years ago, and long after the centromere lost its function.
The other visible footprint of the fusion is the presence of telomeric sequence at the place where the ancestral 2a and 2b telomeres joined. The fusion site was localized by Jacob Ijdo and collaborators in 1991 as they were surveying sequence data for a distinctive six-base-pair repeat sequence commonly found near telomeres.
“The inverted arrangement of the TTAGGG array and the adjacent sequences, which are similar to sequences found at present-day human telomeres, is precisely that predicted for a head-to-head telomeric fusion of two chromosomes.”—Jacob Ijdo and coworkers
Ijdo and coworkers showed that the chromosome 2 sequence had inverted repeats in a pattern that was shorter than functional telomeres, and they hypothesized that the DNA structure would have been stabilized by deletion of the longer telomeric segments after they joined together.
Dating the fusion
Ancient genetic changes that do not vary in living people, like the chromosome 2 fusion, are hard to date. This fusion happened so long ago that the variation of the region surrounding the fusion site is no different from the average variation of the genome. But the fact that the fusion site joined ancestral telomeres provides an unexpected way to estimate the time of the fusion.
The four base pairs of DNA fall into two types: strong bases—cytosine and guanine—are linked by three hydrogen bonds, while weak bases—adenine and thymine—have only two. Across the genome, substitutions from CG to AT, strong to weak, are just as common as substitutions in the other direction, from AT to CG. But in 2007, a team from Katherine Pollard's laboratory noticed that some DNA sequences that evolved fast on the human lineage show a bias with many more substitutions resulting in CG than AT: that is, a bias favoring weak to strong substitutions. Timothy Dreszer and coworkers set out to understand why this might be, surveying the entire genome. They found an intriguing pattern. If substitutions are clustered, with five or more in a short space of a hundred base pairs, there is the same bias toward CG substitutions—but only in the areas close to telomeres.
Dreszer and coworkers concluded that this clustered substitution bias was likely caused by biased gene conversion. In this process, when two chromosomes recombine, a stretch of DNA is copied from one to the other without reciprocal copying, and with preference for one allele over another. Over time it has become clear that biased gene conversion occurs widely across mammals, and is more strongly related to recombination rate in male than in female individuals. In humans, recombination during meiosis in male individuals happens much more often near the telomeres than in other parts of chromosomes.

The history of biased gene conversion has a striking consequence for human chromosome 2. Like all the other autosomes, chromosome 2 has clusters of GC-biased substitutions near its telomeres. But unlike any other chromosome, it also has a spike of GC-biased substitution clusters in the center, just surrounding the place where the ancestral chromosomes fused. That GC bias—even stronger than seen near the telomeres of most other chromosomes—is there because this region was once near the telomeres of ancestral chromosomes 2a and 2b.
Indeed, Dreszer and coworkers recognized, for these GC-biased clusters to exist, there must have been a long time when the human lineage retained the ancestral chromosomes 2a and 2b before they fused together. The team did a quick calculation, estimating that the fusion happened around 740,000 years ago, with a large confidence interval from zero to 2.8 million years.
Last year, Barbara Poszewiecka and coworkers followed up on Dreszer and colleagues' approach. The new study was able to use more data from other ape genomes, including bonobos, and tuned the method to look at the entire pattern of biased clusters across the chromosome in windows of one million base pairs, instead of a single long window near the fusion site as Dreszer's team had done. The result shows very clearly the elevated probability of biased CG clusters surrounding the fusion site in humans.

Their approach led Poszewiecka and coworkers to estimate that the fusion happened 900,000 years ago, with a 95% confidence interval of 400,000 to 1.5 million years. But there were some challenges. The bonobo data in principle should arrive at the same estimate, but instead yielded an estimate of 670,000 years and confidence interval from zero to 1.3 million. The authors observed that the bonobo genome assembly is not yet as high quality as the chimpanzee assembly, which may lead to a somewhat inaccurate picture of substitutions. Poszewiecka and collaborators arrived at their estimates of fusion time by a complicated rescaling, allowing for the fact that the chimpanzee and bonobo chromsosome 2a and 2b both include telomeric segments that are not part of the fusion site in humans.
Still, there's no mistaking the high incidence of GC-biased clusters near the fusion site in humans, and no avoiding the conclusion that the fusion must have happened much closer to the end of our evolution than the beginning.
An ancestral bottleneck?
This week, a new paper in Science reports that the genetic variation in today's people may point to an ancient bottleneck in African ancestors. This may sound like a familiar story—after all, it's the same general approach that in the 1980s gave us the “out of Africa” hypothesis for modern human origins. But this new paper has an unfamiliar twist: This bottleneck may have happened more than 800,000 years ago.
The authors, Wangjie Hu and coworkers, developed a new statistical approach to look for changes of population size in deeper time than previous methods. The approach relies on the site frequency spectrum of genetic variants across the genome, which is broadly similar to the method developed by Aaron Ragsdale and coworkers to examine ancient population structure. I took a close look at the Ragsdale work earlier this year. Where the new approach is different is that Hu and coworkers have not focused on structure or partial isolation among ancient groups; they have instead narrowed their approach to consider only ancient changes in population size.

Their findings suggest a major reduction in population size before 900,000 years ago, followed by an expansion more than 100,000 years later. Hu and collaborators note that this time overlaps with the estimated fusion of chromosome 2, which may have become fixed in the small ancestral group. They suggest that this population was the common ancestor of Neandertals, Denisovans, and African human ancestors, before the three diverged from each other around 700,000 years ago.
Does this hypothesis hold water? A wide range of studies have now examined the genetic variation of recent African peoples to try to understand the demography of ancestral humans. Some of them have concluded that ghost populations existed for hundreds of thousands of years, later merging with recent populations. Ragsdale and coworkers earlier this year found the ghost population idea didn't work as well as a structured ancestral population, in which groups were intermittently connected by gene flow. In their analysis, this ancestral structure was established around a million years ago. That raises a problem with the new results from Hu and collaborators. Clearly if ancestral humans had divided into two or more ancestral stem populations by a million years ago, the present-day variation shouldn't look like it came from a bottlenecked population of 1280 individuals for a hundred thousand years of that time.
Something doesn't add up. Population size is not the only factor that can restrict genetic variation; population structure matters also. The two are not independent of each other. If you want to understand ancient population size, you cannot do that without understanding the pattern of gene flow and isolation among groups.
Since Hu and coworkers used some of the same genetic data as Ragsdale and coworkers, they ought to at least arrive at a similar timeline. A close look at the methods of both papers shows that they actually do. What seems like a discrepancy between the million years for population structure and the 800,000 years for the end of a species-wide bottleneck comes down to different assumptions about generation time: Ragsdale and coworkers assumed 29 years; Hu and coworkers 24 years. Their datasets actually show the same pattern, at roughly the same time. This may be 800,000 years ago, or it may be a million.
The important question is whether the pattern of genetic variation reflects diversification from a single ancestral group, or instead an increase of population size from a bottleneck. I imagine that the real story is some of both. Finding the proportion of each is likely to take better genetic data from much earlier in time.
Back to chromosome 2
Whether these immediate common ancestors of Neandertal, Denisovan, and African ancestral humans were a tight bottleneck or not, this is the most recent population in which the fusion of chromosome 2 could have happened. However, the fusion may have been even earlier. Posziwiecka and coworkers assumed a genetic divergence of Pan and Homo five million years ago, and a humanlike rate of substitutions in the chimpanzee-bonobo lineage. Both assumptions are wrong, and better ones would push their estimate of the fusion time as much as 50% older. Considering the range of uncertainty, nine hundred thousand years ago is indeed possible, but 1.2 million, or even 2 million years ago, may be credible also. Homo erectus, Homo antecessor, Homo naledi, or even Homo floresiensis may have shared the human chromosome 2…or maybe they didn't.
Geneticists have long emphasized the importance of chromosome differences to speciation. The evolutionary geneticist Theodosius Dobzhansky coined the term chromosomal speciation, recognizing that some species of fruit flies are separated by chromosome differences, including differences in number. Chromosomal rearrangements may be problems for reproduction for several reasons. They tend to tend to suppress recombination, and this can result in the buildup of deleterious gene combinations and weaken the effectiveness of purifying selection. Some chromosomal rearrangements can also directly impede sorting of chromosomes during meiosis. This is one of the ways that hybrid sterility between species can evolve.
Despite such potential problems, chromosomal rearrangements nonetheless do evolve. Some are adaptive, for example, if a gene's new location changes its expression in a beneficial way. Many have no effect on survival or reproduction, and may become common or fixed by genetic drift. Even some deleterious chromosomal changes can become fixed by drift in small populations.
The fixation of such changes is common enough that they often separate closely related species. Yet over the last two decades, genomic evidence from many organisms has shown that genetic introgression often does happen between species that differ in chromosome number. Probably the best known example of hybrid sterility is between horses and donkeys, two species that differ in karyotype. But recent work by Hákon Jónsson and coworkers has documented ancient gene flow between several species of equids despite differences in chromosome number and structure. The lesson across many studies is that chromosome incompatibilities are not so simple as once thought.
That may be a useful lesson for fossil hominins. A number of studies have found evidence for so-called “superarchaic” introgression into the genomes of ancient individuals. The most recent research on this subject by Alan Rogers, Nathan Harris, and Alan Achenbach points to a contribution to the common ancestors of Denisovan and Neandertal genomes from a population that separated from our lineage more than 1.9 million years ago. These authors speculate that such a superarchaic ancestor may be a descent of early Homo erectus, as represented at Dmanisi, Georgia. It is possible that this population retained the ancestral karyotype with 48 chromosomes. It will take either a better estimate of the fusion age or ancient DNA from other hominins to say for sure.
It may be that the chromosome 2 fusion was a beneficial change that brought important changes in gene expression to our ancestors. Or it may not have had any immediate functional consequences at all. Whichever is the case, this change is a marker of recent common ancestry of today's people and known ancient genomes. Discovering the fossil population that had this change might give us clues about how these ancient humans succeeded.
Notes: My post describing the study from Ragsdale and coworkers, “Ghostbusters of human origins” has a lot more detail on the analysis of population structure and alternative views about population models.
In an earlier draft of this post I included more detail about how we might get a more accurate estimate of the chromosome 2 fusion. One part of this is understanding the slowdown of human mutation rate described by Søren Besenbacher and collaborators in a 2019 paper. The broader implications are fleshed out in a review by Manjusha Chintalapati and Priya Moorjani in 2020.
There's much more to say about the importance of chromosome rearrangements in evolution and speciation. It's a deep literature and while it's clear that such changes correlate with speciation in a broad sense, there is little consensus about their importance as causes of isolation versus effects of other isolating mechanisms. A broader discussion of this in hominins will have to wait for another post.
References
Ayala, F. J., & Coluzzi, M. (2005). Chromosome speciation: Humans, Drosophila, and mosquitoes. Proceedings of the National Academy of Sciences, 102(suppl_1), 6535–6542. https://doi.org/10.1073/pnas.0501847102
Besenbacher, S., Hvilsom, C., Marques-Bonet, T., Mailund, T., & Schierup, M. H. (2019). Direct estimation of mutations in great apes reconciles phylogenetic dating. Nature Ecology & Evolution, 3(2), Article 2. https://doi.org/10.1038/s41559-018-0778-x
Chiatante, G., Giannuzzi, G., Calabrese, F. M., Eichler, E. E., & Ventura, M. (2017). Centromere Destiny in Dicentric Chromosomes: New Insights from the Evolution of Human Chromosome 2 Ancestral Centromeric Region. Molecular Biology and Evolution, 34(7), 1669–1681. https://doi.org/10.1093/molbev/msx108
Chintalapati, M., & Moorjani, P. (2020). Evolution of the mutation rate across primates. Current Opinion in Genetics & Development, 62, 58–64. https://doi.org/10.1016/j.gde.2020.05.028
Dreszer, T. R., Wall, G. D., Haussler, D., & Pollard, K. S. (2007). Biased clustered substitutions in the human genome: The footprints of male-driven biased gene conversion. Genome Research, 17(10), 1420–1430. https://doi.org/10.1101/gr.6395807
Faria, R., & Navarro, A. (2010). Chromosomal speciation revisited: Rearranging theory with pieces of evidence. Trends in Ecology & Evolution, 25(11), 660–669. https://doi.org/10.1016/j.tree.2010.07.008
Hillier, L. W., Graves, T. A., Fulton, R. S., Fulton, L. A., Pepin, K. H., Minx, P., Wagner-McPherson, C., Layman, D., Wylie, K., Sekhon, M., Becker, M. C., Fewell, G. A., Delehaunty, K. D., Miner, T. L., Nash, W. E., Kremitzki, C., Oddy, L., Du, H., Sun, H., … Wilson, R. K. (2005). Generation and annotation of the DNA sequences of human chromosomes 2 and 4. Nature, 434(7034), Article 7034. https://doi.org/10.1038/nature03466
Hu, Wangjie, Hao, Ziqian, Du, Pengyuan, Di Vincenzo, Fabio, Manzi, Giorgio, Cui, Jialong, Fu, Yun-Xin, Pan, Yi-Hsuan, & Li, Haipeng. (2023). Genomic inference of a severe human bottleneck during the Early to Middle Pleistocene transition. Science, 381(6661), 979–984. https://doi.org/10.1126/science.abq7487
Ijdo, J. W., Baldini, A., Ward, D. C., Reeders, S. T., & Wells, R. A. (1991). Origin of human chromosome 2: An ancestral telomere-telomere fusion. Proceedings of the National Academy of Sciences, 88(20), 9051–9055. https://doi.org/10.1073/pnas.88.20.9051
Jónsson, H., Schubert, M., Seguin-Orlando, A., Ginolhac, A., Petersen, L., Fumagalli, M., Albrechtsen, A., Petersen, B., Korneliussen, T. S., Vilstrup, J. T., Lear, T., Myka, J. L., Lundquist, J., Miller, D. C., Alfarhan, A. H., Alquraishi, S. A., Al-Rasheid, K. A. S., Stagegaard, J., Strauss, G., … Orlando, L. (2014). Speciation with gene flow in equids despite extensive chromosomal plasticity. Proceedings of the National Academy of Sciences, 111(52), 18655–18660. https://doi.org/10.1073/pnas.1412627111
Miga, K. H. (2017). Chromosome-Specific Centromere Sequences Provide an Estimate of the Ancestral Chromosome 2 Fusion Event in Hominin Genomes. Journal of Heredity, 108(1), 45–52. https://doi.org/10.1093/jhered/esw039
Poszewiecka, B., Gogolewski, K., Stankiewicz, P., & Gambin, A. (2022). Revised time estimation of the ancestral human chromosome 2 fusion. BMC Genomics, 23(6), 616. https://doi.org/10.1186/s12864-022-08828-7
Rogers, A. R., Harris, N. S., & Achenbach, A. A. (2020). Neanderthal-Denisovan ancestors interbred with a distantly related hominin. Science Advances, 6(8), eaay5483. https://doi.org/10.1126/sciadv.aay5483
Ventura, M., Catacchio, C. R., Alkan, C., Marques-Bonet, T., Sajjadian, S., Graves, T. A., Hormozdiari, F., Navarro, A., Malig, M., Baker, C., Lee, C., Turner, E. H., Chen, L., Kidd, J. M., Archidiacono, N., Shendure, J., Wilson, R. K., & Eichler, E. E. (2011). Gorilla genome structural variation reveals evolutionary parallelisms with chimpanzee. Genome Research, 21(10), 1640–1649. https://doi.org/10.1101/gr.124461.111
Wang, H., Xing, J., Grover, D., Hedges, D. J., Han, K., Walker, J. A., & Batzer, M. A. (2005). SVA Elements: A Hominid-specific Retroposon Family. Journal of Molecular Biology, 354(4), 994–1007. https://doi.org/10.1016/j.jmb.2005.09.085
Yunis, J. J., & Prakash, O. (1982). The Origin of Man: A Chromosomal Pictorial Legacy. Science, 215(4539), 1525–1530. https://doi.org/10.1126/science.7063861