
The contribution of segmental duplications to human diversity
New studies based on long-read sequencing open a new way of looking at variation of these structural variants.


Genomes of people around the world are much alike at the small scale, when you compare them one nucleotide to the next. But as you open the scope to wider sections of chromosomes, people sometimes have large portions that are duplicated, inverted, or deleted. A large chunk of one chromosome may closely match part of another, evidence of translocation from one place to another sometime in their history. These kinds of genetic variations are called structural variants.
Human geneticists are still working on precise counts of structural variants across people and populations. What may seem like a simple matter of counting is in reality one of the frontiers of human genomics.
In this post I'll focus on some new work on one category of structural variant: segmental duplication. These are long sequences that are repeated within a chromosome, or across chromosomes. A new paper out last week by Hyeonsoo Jeong and coworkers confirms that around 6% of the human genome is composed of segmental duplications, and many vary in both the number and locations of copies across chromosomes. What do these newfound variations mean for understanding human evolution and gene function?

Small and big differences
When it comes to genomes, the smallest differences are easy to measure, and large differences can be much harder to find. For most of the last 20 years, the work of sequencing has been built upon short-read technologies. These approaches break a genome into pieces a few hundred base pairs in length, sequence every last bit, and then use computers to align all of the short pieces to the human genome draft.
Short-read approaches work really well across the majority of the genome where most people's chromosomes can be aligned well with each other. Researchers have catalogued millions of small DNA differences between people's genomes, including single nucleotide variants (SNVs) and short insertion and deletion mutations. These small DNA differences—especially SNVs—are essential tools in human genomics. They have enabled researchers to examine the connections of ancient populations with DNA, and to study the correlation of SNVs and phenotypes across the genome.
But short-read approaches start to falter when DNA is repetitive or duplicated, or when differences are much longer than a hundred base pairs. Large-scale insertions, duplications, and structural variations in genomes have been hard to study. A whole gene can move from one place to another, and short-read approaches might miss it entirely.
Ever since the first draft human genome in 2001, specialists in human genomics have been working to fill these gaps in understanding. Structural variants and duplications are potentially very important to genome function. Many of them affect phenotypes within human populations and differences between humans and other species. These large-scale variants also appear in somatic cells and impact the development of cancers.
In the last several years, long-read sequencing technologies have advanced greatly. These approaches, pioneered by companies like Pacific Biosciences (PacBio) and Oxford Nanopore, generate single-pass sequences of 10,000 base pairs or more in length. Those reads can be long enough to span duplications and structural variations, or at least to reveal accurately which SNVs mark the different copies of a duplicated sequence in a genome. These are helping geneticists to build a much better understanding of genome structure and variation.
Recent studies designed to identify structural variants across samples of human genomes have found that more than 10% of the genome is home to structural variants. While many are only found in a handful of individuals, some are common. Structural variants are concentrated in some parts of chromosomes, especially near telomeres and centromeres, with some hotspots interspersed in other areas that may have functional relevance.
Segmental duplications
The new work by Jeong and collaborators is part of this process of uncovering the pattern of structural variants across the genome. The work comes from Evan Eichler's group at the University of Washington, with important contributions from the Jackson Laboratory for Genomic Medicine in Connecticute. The paper is triple-co-first-authored with Jeong, Philip Dishuck and DongAhn Yoo. The main story is that the team has applied long-read sequencing to a sample of 85 people, including some from African populations and some from other parts of the world, finding most of the segmental duplications across their genomes.
The scope of segmental duplications across the genome is substantial. Many of them are concentrated near telomeres or centromeres, areas where structural mutations and exchange of DNA across chromosomes are very common. Others are interspersed through regions that are rich in gene content, leading to duplications of single or multiple genes. They are heterogeneous in size and patterning. Some amount to tens of thousands of base pairs, others much smaller. In some parts of the genome the duplications are highly clustered, while elsewhere they occur in small numbers.

Figure 4 in the paper is a great illustration of just how different in pattern the duplicated regions are. The first panel in this figure, at top right, shows a 100-kilobase segment of chromosome 5, inverted in an individual and covering a handful of duplications. Third from the left on the top row is a densely-clustered region, in 50 kilobases of the cluster has been duplicated again in one individual.
“Although fundamentally different in nature, the number of variable nucleotides in this 6% of the genome in these 85 individuals is comparable to the estimated 84.7 million single-nucleotide polymorphisms discovered genome-wide from sequencing the 2,500 individuals from the 1000 Genomes Project.”—Hyeonsoo Jeong and collaborators
Looking through the examples, it's so clear how challenging these areas were to map without the massive application of long-read sequencing to samples. This approach is really opening a new window to understand genome variation across people and populations.
Variation in Africa and elsewhere
The research shows that African people have more variation in segmental duplications than populations elsewhere. That higher variation in Africa is accompanied by a bias toward higher copy number for duplicated genes.
For example, the paper includes this figure showing the estimated distribution of copy number for 22 gene families in non-African and African samples, respectively.
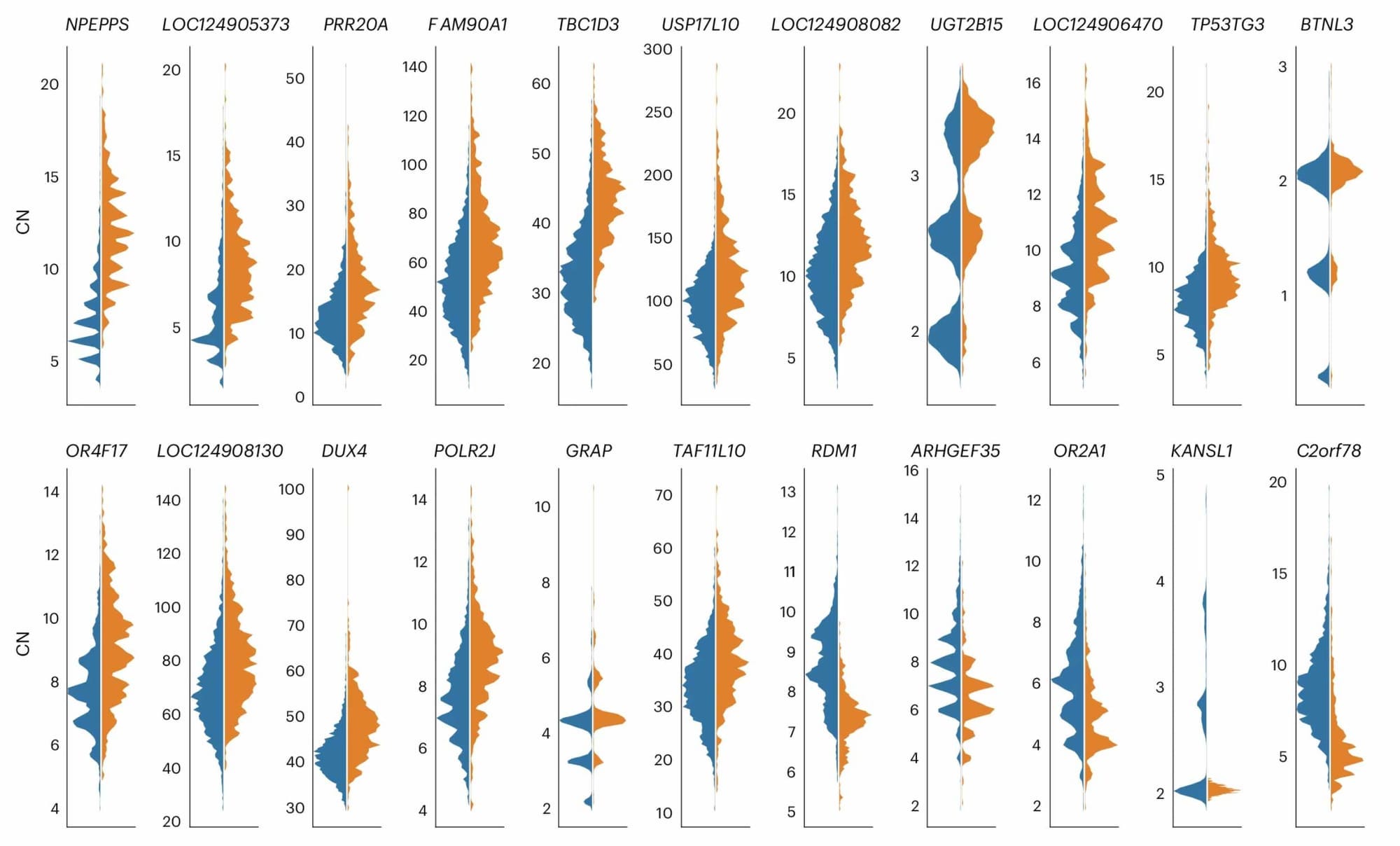
The examples are not all one-way. In a few cases genomes of people outside Africa average higher copy number than African genomes. But across gene families more have a bias toward higher copy numbers in Africa.
Higher variation in Africa by itself is not news. Across all categories of genetic variation, African populations consistently have more variation than non-African populations. That signature is the evidence of a founder effect in the history of non-African populations. And Jeong and coworkers observe that the excess variation in Africa is not out of line when compared with other kinds of variation. Single nucleotide variants, for example, tend to have around 20% higher heterozygosity in Africa. So genetic drift associated with the founder effect shared by non-African groups might explain much of the difference.
But genetic drift should not be biased in direction. Drift can either increase or decrease the number of copies of any given duplicated region, but it does so without preference. Across the entire class of segmental duplications, the average copy number should stay more or less the same.
That's not what Jeong and collaborators found. Instead they found a bias with more copies in Africa. Either populations outside Africa reduced in numbers across segmental duplications, or African populations increased. Either possibility suggests some selection was at play.
What Jeong and coworkers suggest is that selection may have favored increased copy number in some functional domains through much of human evolutionary history.
“Overall higher copy number for duplicated gene families, especially those related to environmental interaction (for example, drug detoxification, immunity), may have provided ancestral human populations with increased genetic diversity in terms of duplicated genes, allowing for selection to operate on different copies to evolve new or modified functions and, therefore, increased fitness.”—Jeong and collaborators
The diversity of environments in Africa coupled with larger population sizes may have favored diversification of gene functions or selection for polymorphism in transcription levels for some gene families. The researchers cite previous work suggesting that the Denisova genome may also reflect higher copy numbers for “many” gene families.
That makes today's non-African populations, with slightly lower copy number across segmental duplications, look like the outliers. A founder effect gave rise to these populations, and in the process they lost variation in a biased way. Stabilizing selection maintaining higher copy numbers in Africa for some of the genes within segmental duplications may have changed in force or direction in the founder population. One reason, as Jeong and coworkers point out, is that unequal crossing-over during recombination sometimes causes problems—and fewer segmental duplications cuts this risk.
Beyond copy numbers
Segmental duplications have other consequences for variation beyond gene copy numbers. Last year, Mitchell Volger and coworkers—also from Eichler's group—showed that sequences found within segmental duplications across the human genome are home to more single nucleotide variants (SNVs) than non-duplicated portions of the genome. The effect is not small: Segmental duplications have 60% more single nucleotide variants than the genome-wide value.
Why should parts of the genome within duplications be more variable at the nucleotide level? One possible explanation is that duplicated DNA mutates faster. Duplications can mismatch during recombination, and this can result in changes to the gene sequence. A common pattern of mismatching recombination causes one allele to replace another on a haplotype, a phenomenon known as gene conversion. By quantifying the effect of gene conversion on the variation of segmental duplications, Volger and collaborators found that around 23% of the single nucleotide variants in duplicated regions may be explained as changes resulting from gene conversion.
The team also considered whether segmental duplications might have more nucleotide diversity because these parts of the genome are older. That might happen if duplicated DNA was maintained by balancing selection, or if background selection was inhibited within these regions. With these processes in play, the DNA within segmental duplications might not mutate faster, it might just have had more time for mutations to build up.
They found that these segmental duplications are indeed older than the average portion of the genome—with estimated time to last common ancestor of 650,000 years compared to 530,000 years for other regions. That increased age was specific to segmental duplications that showed evidence of intergenic gene conversion. These duplications were those that had received “donor” DNA from one or more duplicates in other parts of the genome. The mutational spectrum of SNVs in these duplications, skewed toward guanine and cytosine (GC), also pointed toward the importance of gene conversion as a process. Swapping DNA across genomic locations makes somewhat reduces the force of genetic drift on these segmental duplications.
Measuring genetic differences
It has become conventional shorthand to say that living people's genomes are 99.9% the same. This statement derives from a particular kind of measurement. Sequencing and aligning millions of base pairs from two homologous chromosomes, around 1 in every 1000 base pairs will be a single nucleotide variant between them. That 1 in 1000, a value of 0.001, is a measure of heterozygosity between the genomes, often called the nucleotide diversity.
This is a ballpark figure, and the actual nucleotide diversity varies when comparing different pairs of people. The average within African populations is close 0.001, while the average in populations from other parts of the world is a little lower.
Structural variants are large-scale differences between genomes. A mutation that results in structural variation may change thousands of base pairs at once. Entire genes may be copied again and again, or may be deleted entirely. Like most other kinds of genetic variation, structural variants tend to be individually rare but collectively they add up to a large fraction of the genome. That means that if we could count all the nucleotides that do not match between two homologous human chromosomes, a lot more than 1 in every 1000 base pairs will differ.
As yet, we don't have this accurate count. As noted above, among only the 85 people sampled by Jeong and coworkers, the impact of segmental duplications roughly doubles the number of variable sites that were found to be SNVs in a sample of 2500 people.
A common misconception is that the high similarity between people's genomes is meaningful on its own. That misconception is shared even by some who teach human genetics. People often say, “Humans are 99.9% genetically identical”, expecting that statement automatically conveys the idea that whatever small differences exist cannot matter much.
To see how that is a misunderstanding of concepts, we just have to look at one very common genetic difference. Just over half of people carry two X chromosomes while others carry one X and one Y chromosome. The X chromosome averages around 155 megabases in length, the Y only 60 megabases. Genetic male and female individuals therefore differ in genome length by an average of 95 megabases, an estimated 1.5% of the entire genome. Most genetic male and female individuals do differ in a number of phenotypes, and developmentally what matters to many of those differences is a small region of the Y chromosome including the SRY gene. The count of nucleotides that are different between genomes does not by itself answer the kinds of questions that most people ask.
Counting differences between gene sequences doesn't mean anything intrinsic about the phenotypic differences between people: how different people look, develop, or behave. Many genetic changes make no differences to development, anatomy, and behavior at all. When mutations matter to function, purifying selection tends to weed them out. For example, the average site heterozygosity across protein-coding parts of the human genome is around 0.00042, roughly half the value for noncoding parts of the genome.
What, then, does a measurement of genetic variation actually mean for a population? Such measurements as nucleotide diversity carry information about evolutionary history, but only in combination with other insights.
For example, take the commonly-cited nucleotide diversity in humans of around 0.001 per base pair. One way to add meaning to this measurement is to think about how long this level of heterozygosity must have taken to evolve. By sequencing the genomes of thousands of parents and their children, geneticists have estimated the average rate of single nucleotide mutations across the parts of the human genome without structural variants: 1.2 mutations in 100,000,000 base pairs per generation, or around 1.2 × 10−8 per base pair per generation. Using that information we can estimate that the average difference of 0.001 between two genomes would take around 40,000 generations to build up, if there were no selection on them.
There is not yet a good estimate for the probability of a segmental duplication occurring between parent and child. These events result from such heterogeneous causes that they may not be well summarized by a single rate. Still, measurements of the amount of DNA that differ between two genomes due to structural variants are starting to give valuable insight into the evolution of human ancestors.
Looking forward, the Human Pangenome Project is working to develop a draft genome that incorporates structural variants and other aspects of variation across people's genomes. As that work proceeds, it will become more and more possible to say exactly how many nucleotides, protein-coding genes, microRNA genes, and other kinds of structures vary between people.
Notes: The estimate of structural variation across the genome and distribution of structural variants comes from work by Ebert and coworkers (2021) and Sudmant and coworkers (2015). Average site heterozygosity (nucleotide diversity) for protein-coding sequences is taken from Tennessen and coworkers (2012).
The Human Pangenome Project is an attempt to create a genome reference for humans that recognizes variation across many genomes. This includes building in a map of structural variants. More information about this project can be found in the description by Ting Wang and collaborators from the Human Pangenome Project Consortium.
References
Ebert, P., Audano, P. A., Zhu, Q., Rodriguez-Martin, B., Porubsky, D., Bonder, M. J., Sulovari, A., Ebler, J., Zhou, W., Serra Mari, R., Yilmaz, F., Zhao, X., Hsieh, P., Lee, J., Kumar, S., Lin, J., Rausch, T., Chen, Y., Ren, J., … Eichler, E. E. (2021). Haplotype-resolved diverse human genomes and integrated analysis of structural variation. Science, 372(6537), eabf7117. https://doi.org/10.1126/science.abf7117
Jeong, H., Dishuck, P. C., Yoo, D., Harvey, W. T., Munson, K. M., Lewis, A. P., Kordosky, J., Garcia, G. H., Yilmaz, F., Hallast, P., Lee, C., Pastinen, T., & Eichler, E. E. (2025). Structural polymorphism and diversity of human segmental duplications. Nature Genetics, 1–12. https://doi.org/10.1038/s41588-024-02051-8
Sudmant, P. H., Rausch, T., Gardner, E. J., Handsaker, R. E., Abyzov, A., Huddleston, J., Zhang, Y., Ye, K., Jun, G., Hsi-Yang Fritz, M., Konkel, M. K., Malhotra, A., Stütz, A. M., Shi, X., Paolo Casale, F., Chen, J., Hormozdiari, F., Dayama, G., Chen, K., … Korbel, J. O. (2015). An integrated map of structural variation in 2,504 human genomes. Nature, 526(7571), 75–81. https://doi.org/10.1038/nature15394
Tennessen, J. A., Bigham, A. W., O’Connor, T. D., Fu, W., Kenny, E. E., Gravel, S., McGee, S., Do, R., Liu, X., Jun, G., Kang, H. M., Jordan, D., Leal, S. M., Gabriel, S., Rieder, M. J., Abecasis, G., Altshuler, D., Nickerson, D. A., Boerwinkle, E., … ON BEHALF OF THE NHLBI EXOME SEQUENCING PROJECT. (2012). Evolution and Functional Impact of Rare Coding Variation from Deep Sequencing of Human Exomes. Science, 337(6090), 64–69. https://doi.org/10.1126/science.1219240
Vollger, M. R., Guitart, X., Dishuck, P. C., Mercuri, L., Harvey, W. T., Gershman, A., Diekhans, M., Sulovari, A., Munson, K. M., Lewis, A. P., Hoekzema, K., Porubsky, D., Li, R., Nurk, S., Koren, S., Miga, K. H., Phillippy, A. M., Timp, W., Ventura, M., & Eichler, E. E. (2022). Segmental duplications and their variation in a complete human genome. Science, 376(6588), eabj6965. https://doi.org/10.1126/science.abj6965
Wang, T., Antonacci-Fulton, L., Howe, K., Lawson, H. A., Lucas, J. K., Phillippy, A. M., Popejoy, A. B., Asri, M., Carson, C., Chaisson, M. J. P., Chang, X., Cook-Deegan, R., Felsenfeld, A. L., Fulton, R. S., Garrison, E. P., Garrison, N. A., Graves-Lindsay, T. A., Ji, H., Kenny, E. E., … Haussler, D. (2022). The Human Pangenome Project: A global resource to map genomic diversity. Nature, 604(7906), 437–446. https://doi.org/10.1038/s41586-022-04601-8